Mac Vet Rev 2015; 38 (2): 223 - 232
 10.14432/j.macvetrev.2015.09.055
10.14432/j.macvetrev.2015.09.055
Received: 16 July 2015
Received in revised form: 04 September 2015
Accepted: 10 September 2015
Available Online First: 16 September 2015
Published on: 15 October 2015
Keywords: brucellosis, clinical samples, DNA, real time PCR, MLVA-16
Bacteria from the genus Brucella are causative agents of brucellosis – a zoonotic disease which affects many wild and domestic animal species and humans. Currently, ten Brucella species are officially recognized: B.melitensis, B. abortus, B. suis, B. canis, B. ovis, B. neotomae, B. pinnipedialis, B. ceti, B. Microti and B. inopinata. This classification of the genus Brucella is based on the differences in host preference, pathogenicity, phenotypic traits and genetic structure. However, it is very likely that novel species will be included in the genus soon, mainly as a result of advancement and increased use of molecular diagnostic tools (1).
Infections in animals result in significant economic losses due to induced abortions, infertility, reduced milk and body weight production. Moreover, the presence of brucellosis in animals represents a constant risk for human infections, which are commonly characterized with fever, malaise, sweating and lymphadenopathy (2). In the absence of vaccine for human use, control of human brucellosis remains largely dependent on the control of the disease in animals (3).
Taking into account the significant socio-economic and public health impact of brucellosis, its control is of great importance for endemic areas. Different scientifically approved approaches are available for brucellosis control. However, the chosen control strategy could be successful only if it is adapted to the current epidemiological situation, as well as to the economic and political situation in the country or the region (4, 5). Epidemiology of brucellosis is very complex due to the possible involvement of different mammal (animal) and different Brucella species. This fact highlights the importance of the diagnostic procedures for detection and typing of Brucella, as powerful epidemiological tools which are indispensable for a successful control program.
The only unequivocal confirmation of Brucella spp. infections is laboratory detection and identification of the brucellae in the affected organism. Until recently, isolation of Brucella has been considered as a gold standard assay for brucellosis diagnosis (6, 7). However, significant advancement of molecular techniques and their increased application in the diagnostic laboratories during the past two decades, has led to the formal recognition of these techniques as valid assays for definite diagnosis of brucellosis (8). Unlike isolation, PCR based molecular tests do not require the presence of vital bacteria in the samples, provide quick and objective results and are safe and relatively easy to perform (7, 9). Moreover, due to the possibility for detection of bacterial DNA even in the samples with small number of Brucellae, they have better sensitivity compared to isolation (6, 7, 10). Numerous molecular assays using different targets for Brucella spp. identification have been published in the last twenty five years (11-16). Different methods are available for determination of species (17-21), as well as for typing at subspecies level (22-25). Molecular typing based on the analysis of variable number of tandem repeats analysis (VNTRs) in multiple loci (MLVA), provides valuable epidemiological results and is of great value for investigating outbreaks. MLVA testing scheme consisting of 16 genetic loci (MLVA-16) has high discriminatory power at subspecies level and enable differentiation of unrelated Brucella spp. strains, which could not be differentiated by classical microbiological methods (26-35). The existence of public database with MLVA-16 allelic profiles (http://mlva.u-psud.fr/brucella/) allows comparison of the typed Brucella spp. strains at regional and international level (36). In addition, allelic profiles could be used for species identification or confirmation, if necessary.
The purpose of this study was to establish and evaluate a diagnostic algorithm (flowchart) for Brucella spp. detection and typing, which will exploit the advantages of fluorescence-detection based molecular techniques in terms of sensitivity, safety and testing time and hence provide improved brucellosis diagnostics when clinical samples are tested.
One isolate of B. abortus (field strain MKD-1027), derived from culture collection of the Faculty of Veterinary Medicine-Skopje, and organs from 11 slaughtered seropositive ruminants (Table 3) were used for extraction of genomic DNA. Organs from one animal (spleen, supramammary, iliac and inguinal lymph nodes) were grinded and macerated together in a form of suspension suitable for cultivation, inoculated on selective (Farrell’s) media and archived at -80°C as “tissue sample”. Eleven tissue samples used in the study were randomly selected from the FVMS’s collection of tissue samples kept at -80°C.
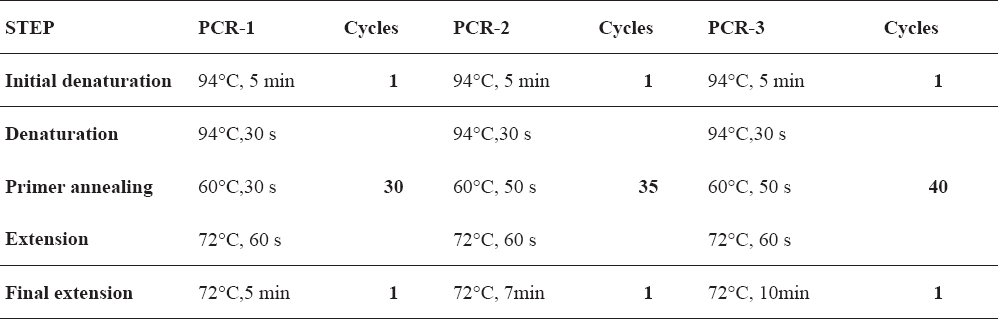
Table 1. Cycling conditions for amplification of VNTR loci for Brucella genotyping
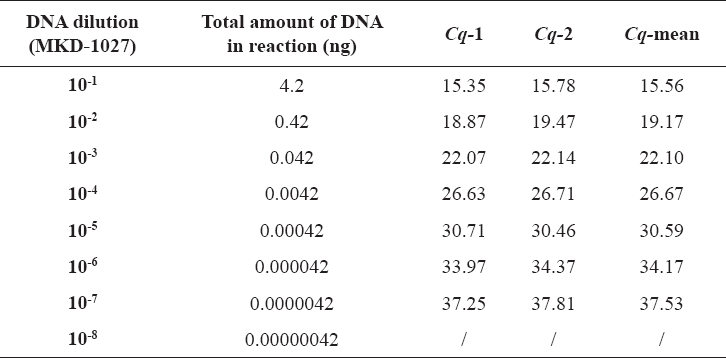
Table 2. Tenfold dilutions of B. abortus (MKD-1027) DNA with appropriate amounts of DNA in reaction and obtained Cq values (tested in duplicate)
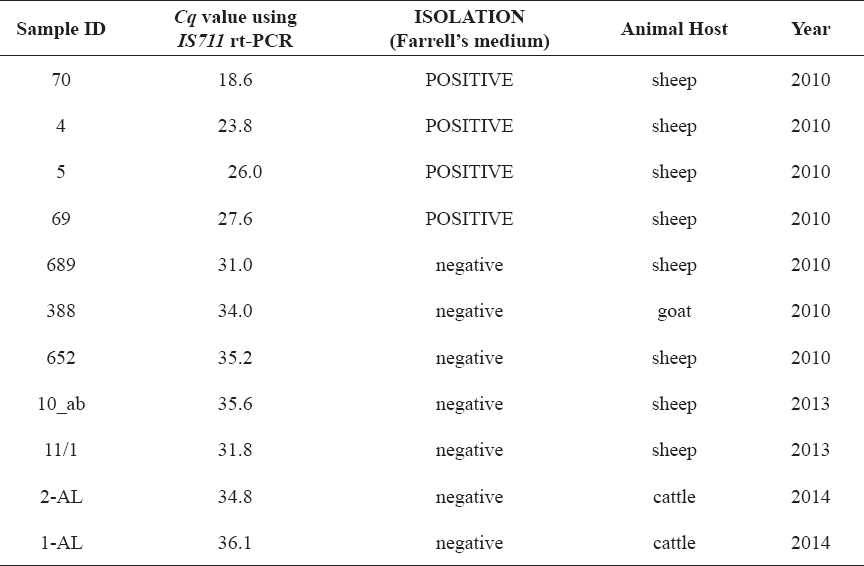
Table 3. Clinical samples in which Brucella DNA was detected by IS711 rt-PCR and MLVA-16 profiles were determined using the described protocols
Pure bacterial DNA used for implementation and optimization of the test protocols for detection and molecular typing was obtained from the MKD-1027 isolate using PureLink Genomic DNA kit (Life Technologies, USA), according to the manufacturer’s instructions for Gram negative bacteria. Concentration and purity of extracted bacterial DNA was determined spectrophotometrically (A260 and A280) using NanoDrop 2000c (Thermo Scientific, USA). Total genomic DNA was extracted from tissue samples using the same commercial kit, according to the manufacturer’s instructions for tissues. The genomic DNA extracted from tissue samples was used to evaluate the performance of implemented testing scheme.
Genomic DNA from field strain MKD-1027 was used for optimization and performance evaluation of implemented real time PCR for Brucella spp. detection. Primers and probe used in the test are targeting IS711 and are described elsewhere (11). Eight tenfold dilutions corresponding to a DNA concentration range between 1.7 ng/µL and 0.17 fg/µL were prepared from the purified DNA sample and tested in duplicate. Real time PCR reaction was prepared in a final volume of 25 µL using 12.5 µL 2x RT PCR reaction mix for probes (Bio-Rad, USA), 8 µL nuclease-free water, 2 µL primer-probe mix and 2.5 µL of DNA sample. Final concentrations of primers and probe were 0.5 µM and 0.2 µM, respectively. Amplification was performed in IQ5 Thermal Cycler (Bio-Rad, USA) under the following conditions: initial denaturation at 95°C for 5 min and 42 cycles of denaturation at 95°C for 15 s, and combined annealing-extension at 60°C for 30 s. Efficacy of the reaction (E), coefficient of determination (R2) and slope of the standard curve were calculated using IQ5 software. One of the DNA dilutions with high quantification cycle (Cq) value has been defined as a future “positive control sample” and was independently tested ten times. Obtained Cq values were used for calculation of coefficient of variation (CV) standing for inter-assay reproducibility and upper and lower limits of the positive control (95% and 99% confidence levels).
For achieving the best analytical sensitivity and repeatability of the capillary electrophoresis system used, all loci from the MLVA-16 panel (22, 24) were amplified in singleplex PCR reactions using 5’ - fluorescently (6-FAM) labeled forward primers and Taq PCR Master mix kit(Qiagen, USA). Depending on the amount of input DNA, three different protocols for PCR reaction set-up and cycling were used (Table 1).
PCR reactions were set in a total volume of 15 µL (PCR-1 and PCR-2) and 25 µL (PCR-3), containing 1 x Qiagen master mix and 0.5 µM of each primer. All PCR amplifications were performed in a TC412 thermal cycler (Techne, UK).
The size of the PCR products was determined using capillary electrophoresis on ABI 310 system (Applied Biosystems, USA). Briefly, undiluted (PCR-3) or diluted (PCR-1, PCR-2) products have been prepared for electro kinetic injection by adding deionized formamide (Applied Biosystems, USA) and GS LIZ500 or GS LIZ1200 (Applied Biosystems, USA) size standards. Amplified loci from referent B. abortus S19 and B. melitensis Rev-1 strains were used as a control strains. Fragment sizing was performed by GeneMapper software ver. 4.0, according to a generated standard curve. Obtained raw data for the size of each locus was recorded in an excel sheet. Measured fragment size was corrected for the previously determined measuring aberration of the instrument. Corrected values were converted in tandem repeat units using Table for allele assignment freely available at MLVA Net for Brucella website (http://mlva.u-psud.fr/brucella/spip.php?article93). Aberration in size measuring was determined for each locus by sequencing. Complete sequences of all 16 loci for referent B. abortus S19 and B. melitensis Rev-1 strains were obtained using ABI Prism Big Dye Terminator (v3.1) cycle sequencing ready reaction kit (Applied Biosystems, USA).
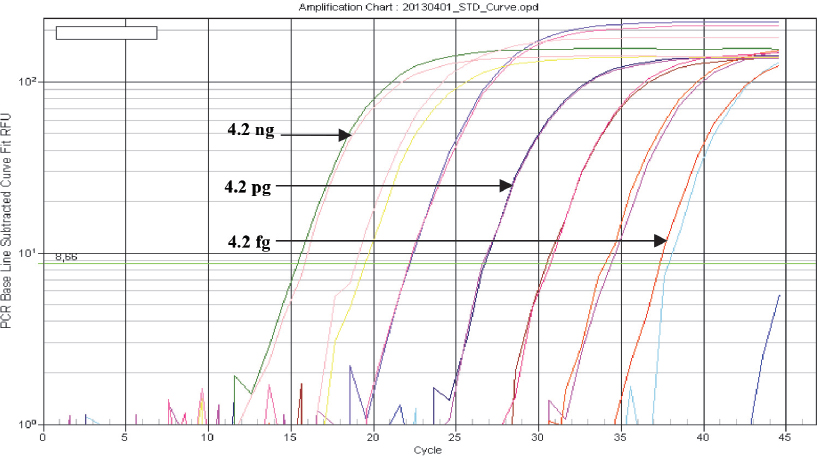
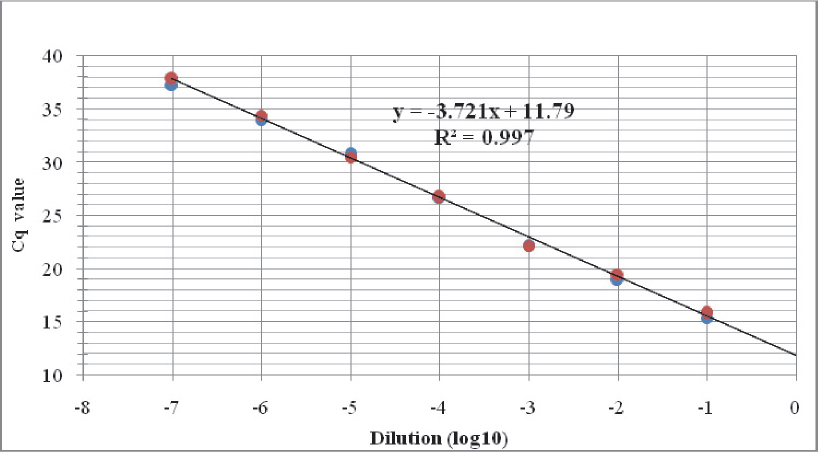
Bacterial DNA was detected in seven out of eight tenfold DNA dilutions when tested with the real time PCR assay for detection of Brucella spp., corresponding to analytical sensitivity of 4.2 fg bacterial DNA (Fig. 1, Table 2). The implemented test protocol amplified broad range of DNA concentrations (4.2 ng to 4.2 fg) with excellent linearity (R2= 0.997) and 85.5% reaction efficacy (Fig. 2).
Figure 1 Amplification curves of B. abortus (MKD-1027) DNA (log view). Tenfold dilutions (from 4.2 ng to 0.42 fg) were tested with the implemented IS711 real time PCR. Fluorescence data is baseline subtracted
Figure 2 Standard curve obtained after testing tenfold dilutions of B. abortus (MKD-1027) DNA with the implemented IS711 real time PCR
Analysis of Cq values obtained in ten independent tests of 10-6 B. abortus (MKD-1027) DNA dilution, indicated very good reproducibility and inter-assay CV of 1.8%. This DNA dilution has been defined as a positive control for all future runs, with upper and lower Cq limits in the following range: 33 – 34.2 (95% confidence level) and 32.5 – 34.7 (99% confidence level) (data not shown).
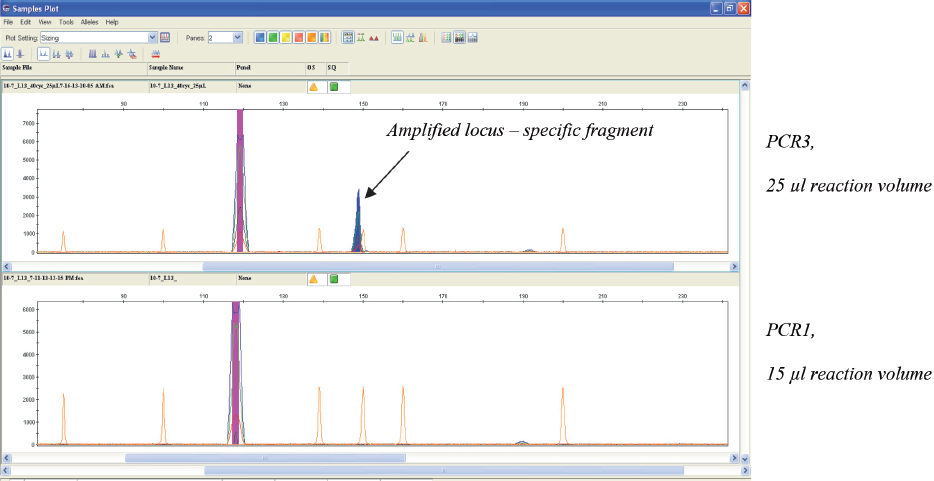
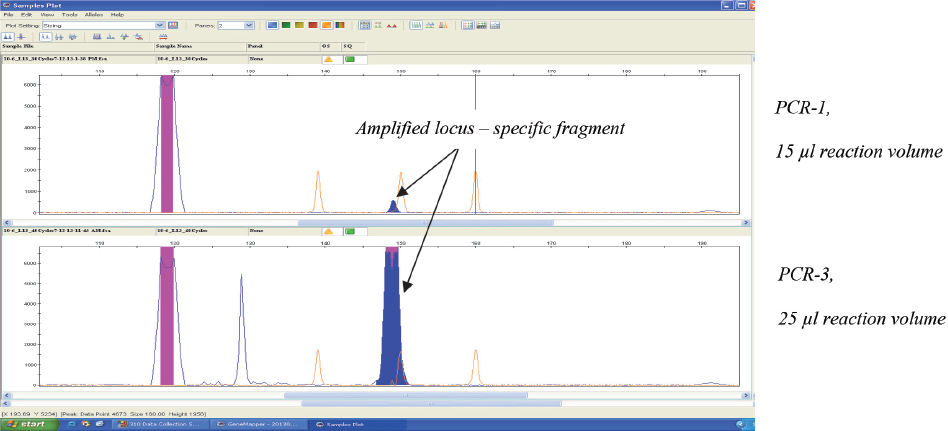
The modified PCR protocol (PCR-3) used for amplification, combined with fluorescence based capillary electrophoresis system for detection, enabled detection of all loci from the MLVA-16 scheme, even in the highest real time PCR positive dilution (10-7) of B. abortus DNA (Fig. 3). Next dilution (10-8) was negative, meaning that the lower limit of detection of the implemented MLVA-16 typing system was equal to the lower limit of detection observed in the implemented real time PCR for detection of Brucella spp. However, PCR-3 protocol should be used only for samples with minor DNA amounts (close to the lower limit of detection), which could not be genotyped using agar-gel electrophoresis. Otherwise, unspecific fragments are likely to appear (Fig. 4), especially if DNA is extracted from clinical samples. The appearance of unspecific fragments could compromise the interpretation of results in situations when their size is very close to the size of existing alleles.
Figure 3 Electropherograms of the products obtained after two different PCR amplifications of bruce30 locus in highest detectable dilution (10-7) of B. abortus DNA. Amplified locus was detected only with the modified (PCR-3) protocol (upper electropherogram)
Figure 4 Electropherograms of the products obtained after two different PCR amplifications of bruce30 locus in 10-6 dilution of B. abortus DNA. Amplified locus was detected with both PCR protocols, but unspecific fragments were present when modified (PCR-3) protocol was used (lower electropherogram)
Brucella spp. DNA was detected in the eleven samples of genomic DNA extracted from the tissues. Obtained Cq values varied, indicating different Brucella concentrations in the tested samples (Table 3). Brucella DNA was detected in seven tissues from which Brucella could not be cultivated, although (as expected) with higher Cq values. Finally, MLVA-16 profiles were successfully obtained for all samples. There were some reproducibility problems when sample with highest Cq value (1-AL) was tested – some of the loci required more than one attempt for obtaining visible signal. This could be explained with low amount of DNA in the 1-AL sample, which was very close to the lower limit of detection of the system.
Confirmation of Brucella spp. infections is very important for successful control of brucellosis in animals. This is especially evident when the eradication programme is in its late stage and the prevalence of disease significantly decreased (4). In this case, rapid confirmation of suspected infections and successful trace-back of its origin and routes, provide significant support to progress towards complete disease eradication. Hence, the selection of proper laboratory methods for rapid, sensitive and specific detection of Brucella spp., as well as highly-discriminatory methods for its typing, play important role in the system of eradication.
As previously reported (11), real time PCR targeting IS711 segment of Brucella spp. is a highly sensitive assay and a good candidate for direct testing of clinical samples. In our study, analytical sensitivity of the IS711 real time PCR assay was 4.2 ng, which is comparable with findings of other authors (11, 37, 38) and proves the robustness of the implemented assay. Theoretically, if we take into account average genome sizes of Brucella members, IS711 real time PCR assay could detect only few bacterial cells. The excellent lower limit of detection is a result of application of real time PCR technology, as well as of the presence of more IS711 copies throughout the Brucella spp. genome.
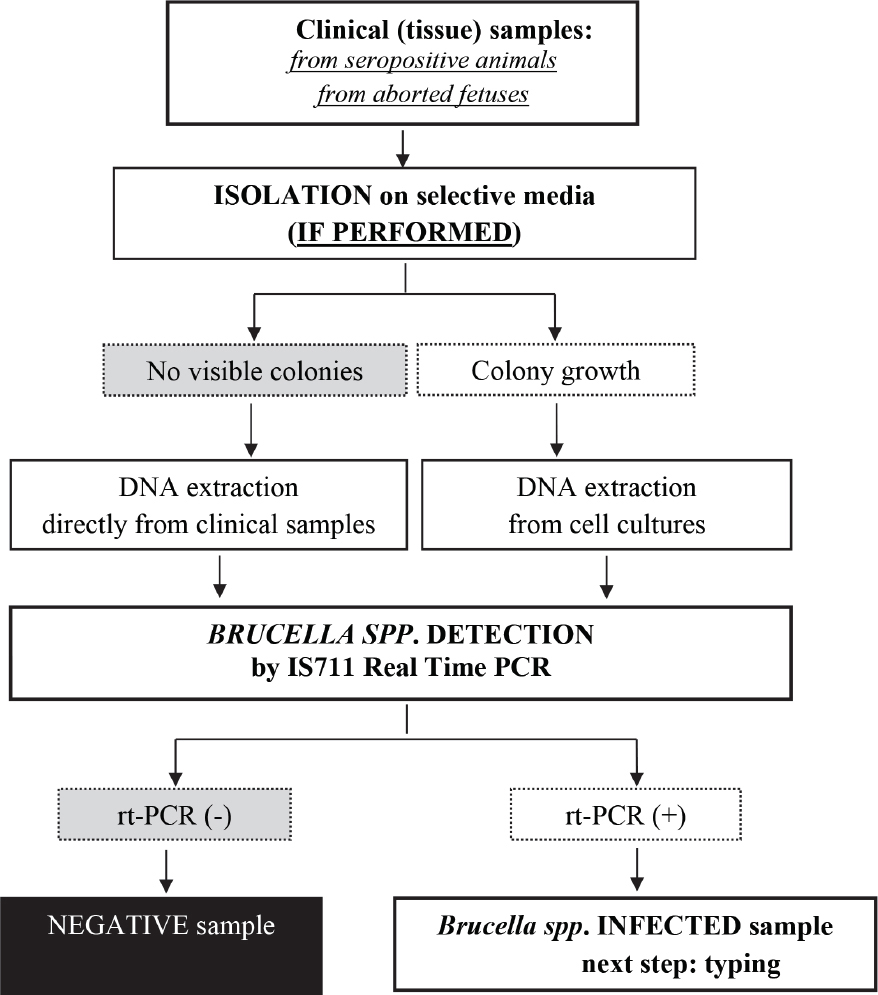
Although the study was not designed to compare the performances of molecular detection and isolation, it confirmed the superior Brucella spp. detection capability of the IS711 real time PCR, compared to the isolation when different clinical samples were tested (Table 3). Our results support the conclusion of other authors (39, 40), that isolation itself is not sufficient for confirmation of infected animals during epidemiological investigations. Therefore, we have established a diagnostic flowchart for Brucella spp. detection/identification based on real time PCR, in addition to isolation on selective media (Fig. 5).
Figure 5 Diagnostic flowchart for Brucella spp. detection
Whenever Brucella spp. is isolated, all molecular tests for species determination and subspecies typing usually perform well, because a high concentration of bacterial DNA is extracted from colonies. In other words, the analytical sensitivity of molecular methods is not an issue when Brucella spp. has been isolated from clinical samples. However, if Brucella spp. is not isolated, but is present in the clinical samples in small numbers which are detectable by the IS711 real time PCR, species and genotype of detected strain could be determined only if highly sensitive technologies are implemented.
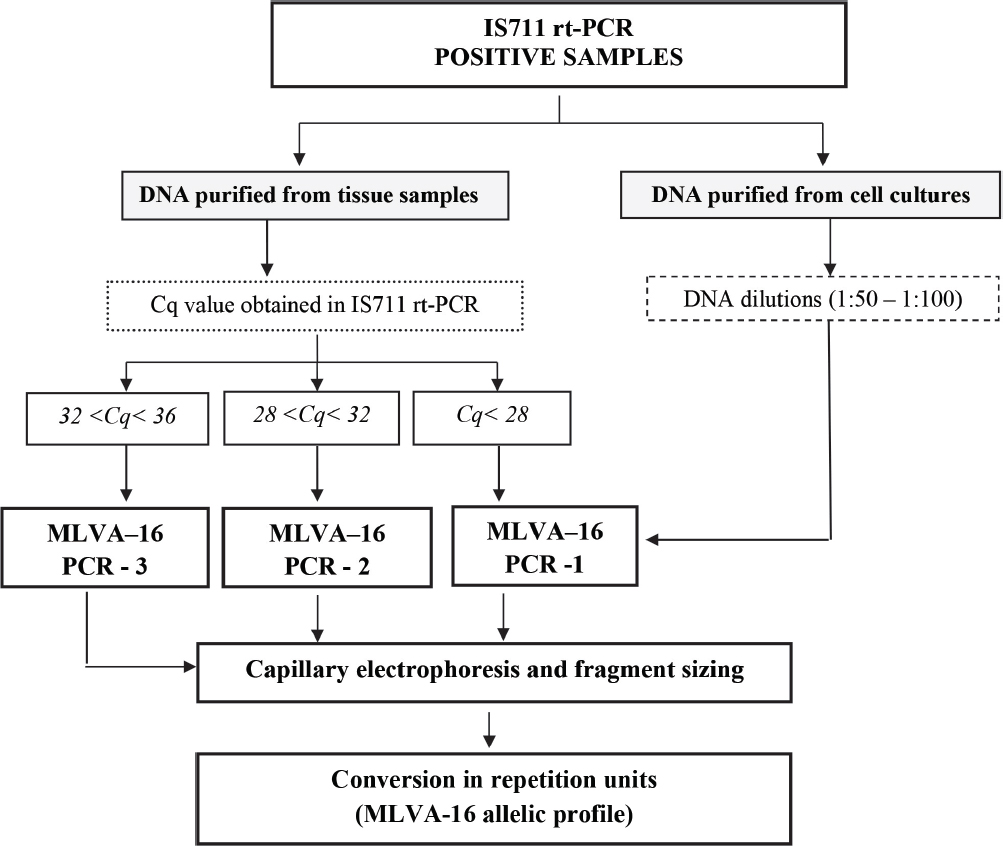
The MLVA-16 typing system that we implemented had equal analytical sensitivity with the above mentioned real time PCR for Brucella spp. detection (Fig. 3), meaning that MVLA-16 allelic profiles could be obtained for all detected Brucella spp. strains. This theoretical conclusion was practically evaluated with the successful genotyping of all clinical samples from Table 3. Moreover, quantitative capabilities of the real time PCR technology provide precise quantization of Brucella DNA loads in the clinical samples, which is very important for selection of the most appropriate MLVA PCR protocol and successful genotyping. Taking into account that DNA sample obtained from the infected tissue contains huge amounts of host DNA, concentration of Brucella DNA could not be measured using the common spectrometric method.
As a summary of all results from the study, we established a diagnostic flowchart for molecular typing of Brucella spp. strains (Figure 6) detected using the laboratory approach given in Figure 5.
Figure 6 Diagnostic flowchart for genotyping detected Brucella spp. strains using MLVA-16
The described testing strategy enables highly sensitive Brucella spp. detection, and successful molecular characterization of detected strains. Its application could improve the effectiveness in infection confirmation and provide significant contribution in the accurate evaluation of epidemiological situations.
Acknowledgment
The authors would like to acknowledge the Joint FAO/IAEA Division and Technical Cooperation Programme of IAEA for the continuous scientific and professional technical support, contributing to the successful upgrading of existing and implementation of new molecular techniques at FVMS.
Copyright
© 2015 Krstevski K. This is an Open Access article distributed under the terms of the Creative Commons Attribution-Non Commercial License (http://creativecommons.org), which permits unrestricted non-commercial use, distribution, and reproduction in any medium provided the original work is properly cited.
Conflict of Interest Statement
The authors declared that they have no potential conflict of interest with respect to the authorship and/or publication of this article.
Citation Information
Macedonian Veterinary Review. Volume 38, Issue 2, Pages 223-232, p-ISSN 1409-7621, e-ISSN 1857-7415, DOI: 10.14432/j.macvetrev.2015.09.055, 2015