

INTRODUCTION
The frequency of outbreaks of food-borne infection cases worldwide is still extremely high (1) in both the Western world and Third World countries (2). Food pathogens are commonly found in the intestines of healthy food-producing animals, and can be transmitted to humans through contamination in the food chain. Thus, a strict control is required of the whole food chain aimed at enforcing contamination detection measures.
Molecular methods for detection and identification of food pathogens have significant benefits as compared to traditional methods due to speed, sensitivity, specificity and accuracy. On the other hand, they often require dedicated instrumentation and higher labour costs. Most molecular methods include an in vitro amplification of nucleic acid via PCR, which is still the most widely applied technique in clinical laboratories. PCR based methods are used to detect, identify and quantify either pathogens or beneficial populations such as fermenting microbes or probiotics (3, 4). An advanced real-time PCR based technique has been approved as suitable and convenient diagnostic technique for a wide spectrum of targets in food-borne pathogen detection and can be performed using a variety of instrumentations including capillary equipment (LightCycler 2.0) and plate-cyclers like LightCycler 480 (5, 6). Multiplex real-time PCR involves simultaneous amplification of more than one locus and thus allows detection of multiple microorganisms in a single reaction. However, the primer sets should be designed with a similar annealing temperature, while providing a method to distinguish between amplicons following thermal cycling (7). It has also been suggested that performing a single, multiplex assay instead of several singleplex analyses might reduce the total costs for testing (8).
The objective of the present study was to provide a specific protocol to detect simultaneously food contamination by any of the five food-borne pathogens, namely Campylobacter jejuni, Mycobacterium bovis, Enterobacter sakazaki, Shigella boydii, Clostridium perfrigens. The developed assay can be used to analyse any kind of food, including but not limited to milk, cheese and meat products. The performance of this assay was assessed by using DNA purified by ATCC strains.
MATERIAL AND METHODS
Samples
The lyofilized Shigella boydii and Enterobacter sakazaki strains were aerobically grown in a Brain Heart Infusion Broth (Oxoid, Italy) at 37°C for 24 hours. Campylobacter jejuni strain was grown in a Brain Heart Infusion Broth (Oxoid, Italy) under a microaerophilic atmosphere (CO2Gen, Oxoid, Italy) at 42°C for 24-48 hours. Clostridum perfringens was grown anaerobically, using BBL GasPak™ Systems (BD, USA), in a Brain Heart Infusion Broth (Oxoid, Italy) at 37 °C for 48 hours. After overnight incubation from fresh colonies, bacterial cells were collected by centrifugation and subjected to genomic DNA isolation, as previously described (9). The bacterial strains and the isolated genomic DNA are described in Table 1.
Table 1. Reference target bacterial strains used in this study and primers designed using PRIMER EXPRESS (ver. 3.0)
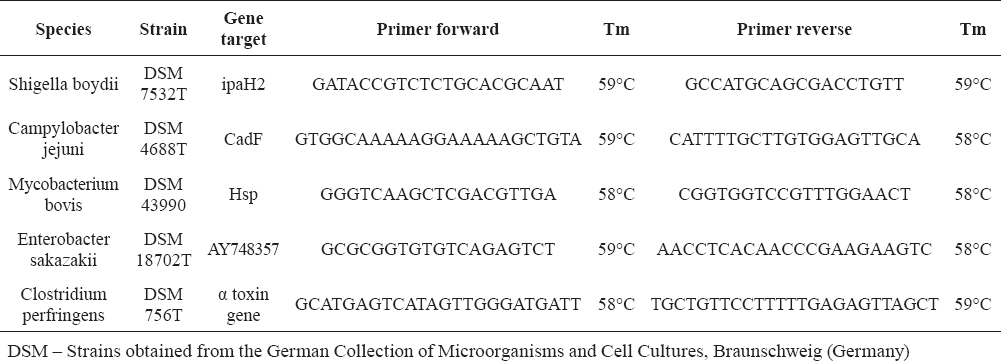
For Mycobacterium bovis only genomic DNA was available. The concentration of DNA used for reference target strains varied from 3.9 to 11.2 ng/ml. Pure and mixed samples were prepared using isolated bacterial DNA and NA-free water from Roche Diagnostics (Germany). No real food samples were used.
Primer design
Primers for real-time PCR amplification were designed using PRIMER EXPRESS (ver. 3.0). Primer pairs were ordered from Tib-MolBiol (Germany) and tested for EvaGreen assays on LightCycler 2.0® (Roche Diagnostics GmbH) using 5x HOTFIREPol® EvaGreen® qPCR Mix Plus (Capillary) with 7.5 mM MgCl2 ready-to use mastermix (Solis BioDyne). Primer sequences and the accession number of target genes are listed in Table 2.
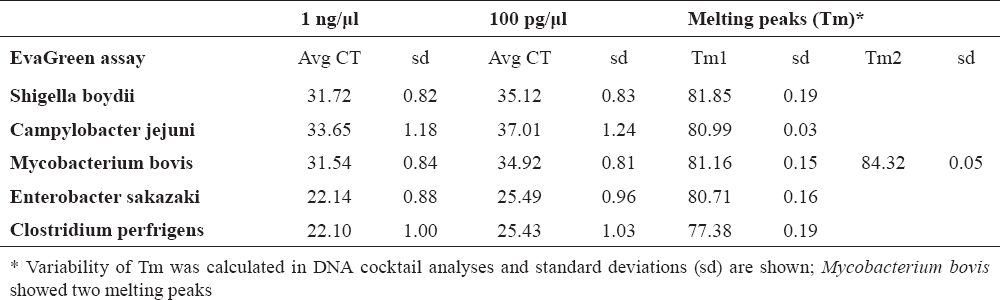
Table 2. Threshold cycle (CT) of quantification analysis obtained in 10-fold dilution of the pure culture genomic DNA (1 ng and 100 pg) and major melting peaks of amplicons
EvaGreen PCR assay
Quantitative PCR reactions followed by melting curve analysis were performed in a final volume of 20 ml containing 2 µl of template genomic DNA, 4 µl of 5x HOT FIREPol® EvaGreen® qPCR Mix Plus (Capillary) with 7,5 mM MgCl2, 5 µl of primer cocktail (0.5 µl of each forward and reverse primer) and 10 ml of molecular grade water. Tenfold dilutions of the target genomic DNA (1 ng/ml and 100 pg/ml) were tested to determine the fair amount of template DNA detected by the assay (PCR sensitivity). The reactions were carried out by using LC Capillaries 20 µl. Each sample was tested in triplicate with the following thermal profile: 95°C for 15 min for hot-start, followed by 45 cycles at 95°C for 15 s, 60°C for 20 s and 72°C for 30 s. The melting point analysis was preceded by a denaturation step 95°C for 5 s and an annealing step 65°C for 15 s, followed by continuous fluorescence recording from 65°C till 90°C 0.05°C/s and a cooling step 40°C for 15 s.
Comparison of two different analysis methods: Tm calling versus Melting Curve Genotyping
The melting curve results at λ 530 nm were analyzed with LightCycler480 SW1.5.0 using two different options: automated Tm Calling and Melting Curve Genotyping to investigate the potential to identify the pathogens via qPCR product melting peak and melting profile. Both analyses were used only for samples with Ct less than 37 in quantitative analysis.
RESULTS
Analytical sensitivity and specificity
In multiplex testing, sensitivity was detected by analysis of tenfold dilution series of bacterial DNA. The detection of 100 pg of pathogen DNA was confirmed (Table 2).
The melting curves for PCR products were analyzed by two methods, while specificity of the multiplex assay was confirmed by testing against all five targets: Campylobacter jejuni, Mycobacterium bovis, Enterobacter sakazaki, Shigella boydii, Clostridium perfrigens. In samples containing artificial mixtures of pathogens DNA, usually no more than two major pathogens were identified by melting curve analysis. However, in all capillaries no false-positive or false-negative results were generated, confirming the excellence of the developed assay (Fig. 1).
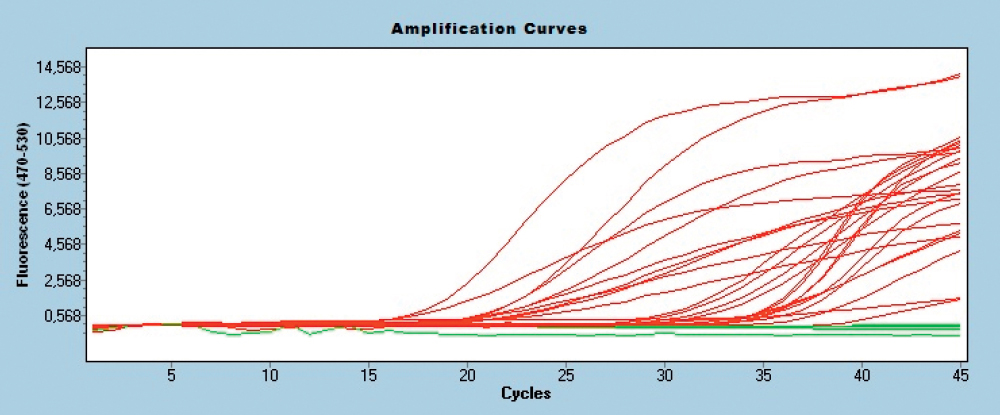
Figure 1. Amplification curves of EvaGreen assays in multiplex test with DNA cocktails
Intra- and inter-assay reproducibility
To obtain values for the intra- and inter-assay variation of the assay, 1 ng and 100 pg of purified genomic DNA was analyzed in triplicate. The coefficients of variation (CV) of the Ct values ranged from 0.5 to 2.1 % for intra-assay experiments and from 2.3 to 4.5 % for inter-assay experiments.
The coefficients of variation of the Tm values ranged from 0.05 to 0.21 % for intra-assay experiments and from 0.06 to 0.25 % for inter-assay experiments.
Tm calling versus Melting Curve Genotyping
The robustness of the assay was evaluated by comparative analysis of PCR product melting curves using different software features – Tm calling and Melting Curve Genotyping (MCG).
Tm calling allowed the identification of PCR products by major product(s) melting temperature, however in MCG the pattern of the melting curve was identified. Tm calling showed clear differentiation of samples with single pathogen genomic DNA as shown in Table 2. In mixed samples (containing DNA from two or more pathogen) the major component was always identified, thus the food would be rejected in any pathogen contamination included in the assay. However, the procedure did not allow identifying the minor components, especially if three pathogens were mixed for analysis.
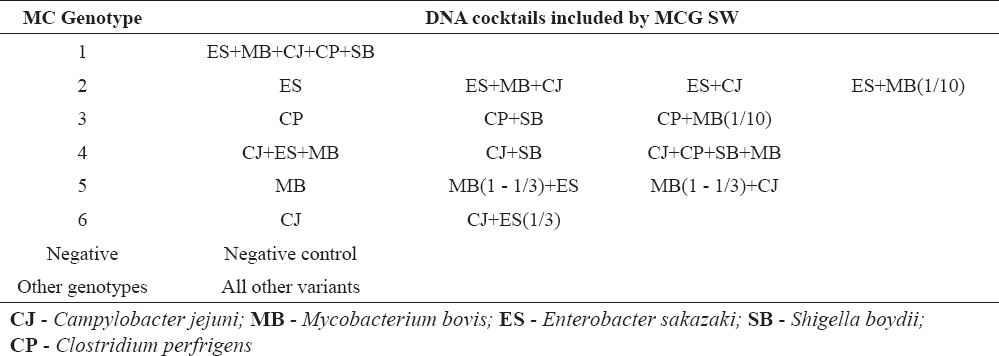
Melting Curve Genotyping showed similar performances, but because the basis for identification is the melting curve pattern, Mycobacterium bovis got the priority for decision. Mycobacterium bovisamplification resulted with products identified with two melting peaks and it fit best under the term ‘pattern’. Thus, Mycobacterium bovis was identified as a major pathogen even in cases where it was in a mixture even in a three times lower concentration than other pathogens. The Melting Curve Genotyping results are shown in Figure 2 and Table 3.
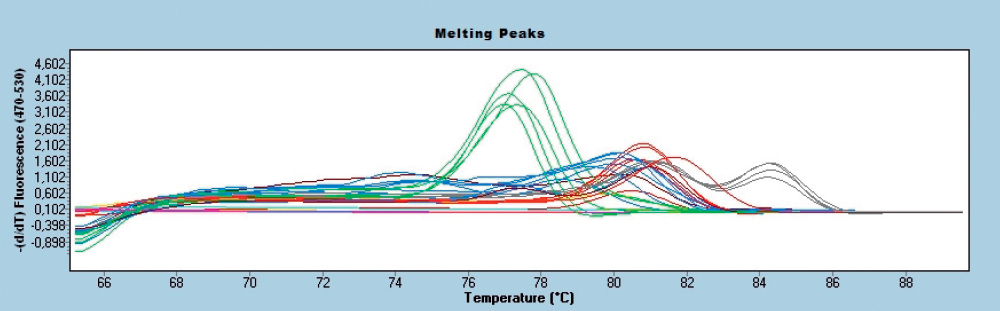
Figure 2. Species identification in melting curves using melting point genotyping feature of LightCycler 480 software for DNA cocktails

Table 3. Melting Curve Genotyping results for different pathogen DNA cocktails
DISCUSSION
It is generally accepted (10), that prevention of food-borne disease basically depends on surveillance and prompt identification of pathogens in food products. The major advantage of the current molecular method is the lower price and the short time necessary to obtain the results.
A crucial step for molecular assessment of microbial communities is the selection of a gene or a genetic marker that can be used to differentiate a wide variety of organisms (11). Usually, the specificity of assays is ensured by using hydrolysis or hybridization probes (3). Indeed, any of all additional chemistries, even the most widely used TaqMan® chemistry, will significantly increase the costs of assay.
Several real-time PCR assays for single reaction have been developed for the detection of the pathogens subjects of our interest and the trend has been moving towards strategies for rapid identification of more than one pathogen through the development of multiple analysis platforms (12, 13, 14, 15).
The real-time PCR assay described in this study has the potential to be a fast screening assay for several pathogens, enabling simultaneous processing of many food samples. Because the assay development does not include the sample preparation steps, the only prerequisite is to obtain good quality (good purity and sufficient concentration) purified DNA from various samples and is not limited to food, but can also be used for HACCP (analysing surfaces, etc.) risk analysis or other goals.
CONCLUSION
In conclusion this assay may be used for accurate and rapid diagnosis of food-borne outbreaks, it has the potential to be used in routine diagnostic laboratories providing a simple, fast, cheap and sensitive alternative method to culture-based or TaqMan qPCR methods.
CONFLICT OF INTEREST STATEMENT
The authors declared that they have no potential conflict of interest with respect to the authorship and/or publication of this article.



