Abstract
Avian avulavirus type 1 (AAvV-1) belongs to the family Paramyxoviridae, genus Avulavirus. Even though belonging to a single serotype, there is great genetic variability between these viruses. On the basis of the available complete fusion (F) gene and whole genome sequences and using the genotype classification system, AAvV-1 viruses are divided in two major groups: class I and class II. Class I viruses are predominantly avirulent viruses and majority of the isolations come from waterfowl and domestic poultry from live bird markets in USA and China although isolations from wild birds are reported globally. In our study we used classical, molecular and phylogenetic tools to characterize an AAvV-1 isolated from a mute swan in Macedonia. Based on the complete F gene sequence, we have concluded that the virus designated as AAvV-1/mute swan/Macedonia/546/2012 (KP123431) belongs to the class I of AAvV-1 with an avirulent cleavage site motif 112ERQER*L117. The virus could not be assigned to any of the four currently existing subgenotypes (1a, 1b, 1c or 1d) of the single genotype 1 of class I viruses. Two distant viruses, isolated from goose in Alaska in 1991 and from goose in Ohio in 1987, shared the highest nucleotide sequence identity of the complete F gene with the isolate in our study: 92.7% and 92.8%, respectively. This is the first report of isolation of class I AAvV-1 in Southeastern Europe. The asymptomatic nature and the underreporting of sequences from the class I viruses impede the understanding of the molecular epidemiology and evolution of this group of viruses.
Keywords: AAvV-1, class I, mute swan, Macedonia
INTRODUCTION
Avian avulavirus type 1 (AAvV-1) viruses, also known as Newcastle disease (ND) viruses (NDV), belong to the family Paramyxoviridae, genus Avulavirus (
1,
2). Although belonging to a single serotype, there is a great genetic variability between these viruses. The present classification system that distinguishes two major AAvV-1 groups, class I and class II, is widely used by the international scientific community (
3). Due to increasing availability of sequencing techniques and sequences included from previously unreported areas, new genotypes/ subgenotypes are being identified frequently (
4,
5). Nowadays, based on the complete F gene sequence, class I consists of one genotype further divided into four subgenotypes (1a, 1b, 1c and 1d), while class II consists of 18 genotypes, some of them further divided into subgenotypes (
4,
5,
6). The presence of viruses that are unassigned to any of the four genotypes (
4,
6,
7,
8), combined with the addition of novel viruses may lead towards an extension of the current classification of the class I viruses by designating new subgenotypes. According to the criteria set for the establishment of a new genotype/subgenotype (
4), these unassigned viruses possess a sufficient evolutionary distance for the designation of new subgenotype but other independent isolations of viruses with separate epidemiological background lack.
Majority of isolations of class I viruses comes from waterfowl and domestic poultry from live bird markets in the USA, China and Hong Kong (
9,
10,
11,
12), although isolations from wild birds are reported in Europe, USA, China, Russia, and Japan (
5,
6,
7). The vast majority of these viruses are avirulent (
8,
9,
11,
12,
13) and because of their low virulence they do not fall under the OIE criteria for declaring a ND outbreak, i.e. no trade restrictions are imposed in the zone or country and the socio- economic impact is negligible (
14). Mutation of low virulent to virulent viruses is a very rare event and this has happened in the past only in two instances for both class I and class II AAvV-1 (
15,
16). To date, the only class I virus reported to be virulent is the one isolated in the Republic of Ireland during an outbreak of ND in domestic poultry in 1990 (
15). It was proven that the cause of the outbreak were few mutations in previously circulating low virulent virus in waterfowl (
17). It was suggested that this could be a very rare event, given the negative selection of the F protein and conserved nature of its cleavage site (
18). However, in an experimental set up an avirulent virus from a waterfowl origin became virulent in chickens after several passages in air sac and brain tissue, by acquiring a virulent cleavage site pattern (
19). This data highlights the need for continuous surveillance for low virulent viruses and use of sensitive diagnostic assays able to detect the diverse genetic makeup of AAvV-1 viruses. Moreover, these studies should be specially designed to detect circulating AAvV-1, as majority of the detections are the result of avian influenza surveillance studies (
20).
The detection of virulent NDV in wild birds as a result of an ongoing outbreak in domestic poultry has been reported in Macedonia (
21). However, detection of low virulent viruses in wild birds has not been reported in the country so far. The aim of this study was to characterize AAvV-1 isolated from a mute swan in Macedonia using classical, molecular and bioinformatics tools.
MATERIAL AND METHODS
Sample data and virus isolation
The virus described in this study was isolated from a mute swan (Cygnus olor) found in the vicinity of the town of Strumica (N 41o26’27. E 22o38’15.) in the southeastern part of Macedonia. The swan demonstrating signs of emaciation and lethargy, was caught on 29 February 2012. On the same day the bird was transferred and quarantined in the zoo in Skopje, where it died a few days later, i.e. on 2 March 2012. Field necropsy was performed and the visceral organs (trachea, lung, liver and kidney) including intestines were sampled and further processed for laboratory investigation at the Faculty of Veterinary Medicine in Skopje. Supernatant of the homogenized organs was inoculated in the allantoic cavity of 11 day old embryonated chicken eggs (ECE) for the isolation of the virus in two consecutive passages according to the international recommended protocol (
14).
Haemagglutination inhibition test
Haemagglutination inhibition (HI) test was performed on the positive haemagglutinating allantoic fluid, harvested separately from the inoculated pool of visceral organs and intestines according to the recommended protocol(
14). Standardpanel ofreference antisera for AAvV serotypes from 1 to 9 (Animal and Plant Health Agency, Weybridge, UK), monoclonal antibody (MAb): 617/161 specific for pigeon variants of AAvV-1 and MAb 7D4 specific for La Sota and F strains (
22), were used for characterization of the isolate. To exclude notifiable avian influenza virus, the allantoic fluid was tested using H5 avian influenza antisera (A/cygnus olor/1/Croatia/2005 H5N1 and A/turkey/Italy/1980 H5N2) and H7 avian influenza antisera (A/turkey/Italy/2732/99 H7N1 and A/turkey/ Italy/8535/2002 H7N3).
Nucleic acid extraction, reverse transcriptase PCR (RT-PCR), real-time RT-PCR
Extraction of viral RNA from the allantoic fluid was done using QIAamp Viral RNA Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. One-step real-time RT-PCR’s using AgPath- ID One-Step RT-PCR kit (Thermo Fisher Scientific, Carlsbad, USA) for the detection of the M gene, F gene of mesogenic and velogenic strains and F gene of the early genotypes were performed (
23), while to exclude avian influenza, M gene assay was done as well (
24). One-step RT-PCR was performed using OneStep RT-PCR Kit (Qiagen) generating a PCR product of 433 bp (
25). To obtain a complete F gene, sequence primers described by Lindh et al., were used (
7). The amplified PCR products were visualized in 1.5% agarose gel stained with ethidium bromide. Clean up of the PCR products was done using PureLink® PCR Purification Kit (Invitrogen, Carlsbad, USA).
Sequencing and phylogenetic analyses
Purified PCR products were sequenced using Big Dye Terminator v3.1 kit (Applied Biosystems, Foster City, USA) with POP-7 polymer (Applied Biosystems) in the ABI Genetic Analyzer 3730 XL (Applied Biosystems). Editing of the sequence data and building a consensus sequence was done using Staden Package version 2.0.0 (
26). Alignment using Clustal W method and phylogenetic analyses using Maximum Likelihood method based on the General Time Reversible (GTR) model were done using MEGA 6 (
27,
28). The GTR model was selected based on the values for Bayesian Information Criterion and corrected Akaike Information Criterion. First, 183 class I sequences published in previous studies and representing all sub-genotypes of genotype 1, the unassigned sequences, and sequences closest to the isolate in this study were downloaded from GenBank. A preliminary analysis including the new isolate was done (data not shown). Based on the results, 85 sequences were selected by omitting sequences from the groups of identical or highly similar sequences and taking into consideration epidemiological data (host/country/year), butproviding that the tree topology is maintained. Complete F gene sequences (n=85) consisting of 1662 positions in the final dataset were used for alignment of the sequences and the phylogenetic interpretation was done using MEGA 6 (
28).
RESULTS
Gross lesions during the field post-mortem examination were empty crop and gizzard, hemorrhages of the proventriculus and inflammation of the pleura and lungs. Isolation of the virus was done on ECE, both from the pools of visceral organs and intestines. Haemagglutinating activity of the allantoic fluid was present after the first passage in both samples. Haemagglutination (HA) titer of the allantoic fluid harvested from the inoculated pool of organs was 1/128 (log27), while HA titer of the allantoic fluid harvested from the inoculated intestines was 1/256 (log2 8). HI test using panel of reference AAvV antisera for serotypes 1 to 9 showed positive result using AAvV-1 antiserum only. HI titer using polyclonal NDV antiserum was 1/512 (log29 in both samples. There was no HI present when using MAb 617/161 and MAb 7D4. Real-time RT-PCR’s for detection of the M gene, F gene of mesogenic and velogenic strains and F gene of the early genotypes were all negative. Amplified PCR products gave specific bands for all four primer pairs covering the full length of the F gene of class I AAvV-1 (Fig. 1) and for the primers detecting partial F gene of class I AAvV-1. Nucleotide sequencing of the cleavage site of the F gene revealed an amino acid motif specific for avirulent isolates of AAvV-1, with leucine at the position 117 and less than three arginine or lysine residues between the positions 113 to 116, 112ERQER*L117 (14). Phylogenetic analysis by constructing a phylogenetic tree was done using the complete F gene sequence. The studied virus clustered in the class I AAvV-1, but could not be assigned to any of the current subgenotypes (Fig. 2). GenBank accession number of the complete F gene nucleotide sequence of the isolate AAvV-1/ mute swan/Macedonia/546/2012 is KP123431.
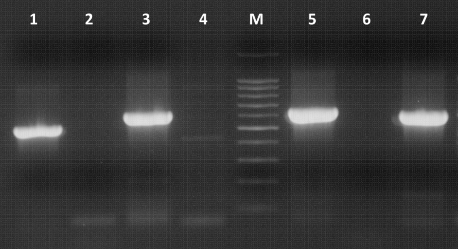
Figure1. Electrophoresis of PCR products amplified using primers designed by Lindh et al., (7). M – molecular marker 100 – 1000 bp, Lane 1 – primers s1 Hf/s1 h, Lane 2 – negative control, Lane 3 – primers s2 Hf/s2 h, Lane 4 – negative control, Lane 5 – primers s3 Hf/s3 h, Lane 6 – negative control, Lane 7 - primers s4 Hf/s4 h

Figure 2. Phylogenetic tree of class I AAvV-1 viruses. The isolate studied here is marked with a black dot. U stands for “unassigned” viruses. The evolutionary history was inferred by using the Maximum Likelihood method based on the General Time Reversible model (
27). Initial tree(s) for the heuristic search were obtained by applying the Neighbor- Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.5672)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 85 nucleotide sequences. There were a total of 1662 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (
28). GenBank accession numbers are shown in parenthesis
DISCUSSION
According to the history, clinical and post- mortem data, the mute swan most probably suffered from an underlying disease not related to isolation of the class I AAvV-1. Post-mortem findings indicating an inability to feed, point to the fact that the bird was sick for a prolonged period of time. Being an avirulent virus (cleavage site 112ERQER*L117), the presence of the virus itself in the organism of the bird most likely was not the reason for its death. Interestingly, the same scenario of isolation of class I virus from Australian white ibis (Threskiornis molucca) that died from an unknown cause was reported in Australia in 2006 (
29). The mute swan was caught near Monospitovo swamp, which is the largest wetland in Macedonia and it is used by various bird species during the nesting period, wintering or as a migration stop. Birds species that are found there include mallard (Anas platyrhynchos), common teal (Anas crecca), wigeon (Anas penelope), garganey (Anas querquedula), black stork (Ciconia nigra), grey heron (Ardea cinerea), little egret (Egretta garzetta) and others (
30). The wetland serves as a sampling site and the time period when the bird was caught corresponds to the intensive activity of volunteer hunters engaged in hunting of wild birds for the purposes of active surveillance of avian influenza. Isolation of the virus was incidental and it likely could have happened geographically at another location or country, since the swan would have gotten sick over a different time period or have flown a different route. To date, reported isolations of class I viruses in Europe are from waterfowl and poultry in Denmark, Finland, France and Germany (
3,
7,
31).
Results from the HI tests clearly demonstrated that the isolate is AAvV-1. The negative result using M-gene assay (
23) is not unexpected, given its low sensitivity to detect class I viruses as a result of at least six mismatches at the probe site sequence (
10).
Phylogenetic analysis of the complete F gene sequence showed that AAvV-1/mute swan/ Macedonia/546/2012 (KP123431) could not be assigned to any of the four currently existing subgenotypes (1a, 1b, 1c or 1d) of the single genotype 1 of class I viruses (
4). Although the virus fulfills the average distance criteria per site (3-10%) for designation of a new subgenotype (
4), it is the only representative in its group, i.e. three more isolates with separate epidemiological background are needed for the establishment of a novel subgenotype, e.g. 1e (
Fig. 2). Two distant viruses isolated from goose in Alaska in 1991 (AB524405) and from goose in Ohio in 1987 (EF564833) share the highest nucleotide sequence identity of the complete F gene with the isolate in our study, 92.7% and 92.8%, respectively. This fact confirms that there is an absence of sequences to fill the epidemiological gap on the global level. These two similar viruses are also not assigned to any of the four subgenotypes, instead they cluster in two separate groups on the phylogenetic tree. There are nine branches of unclassified viruses that can potentially become novel subgenotypes, if more sequences clustering in these branches become available to meet the criterion of sufficient number of epidemiologically independent isolates (
4). Comprehensive phylogenetic data of class I viruses is limited compared to class II viruses (
13,
31), the latter being isolated from more species and diverse geographical locations. Therefore, in order to understand more thoroughly epidemiology of class I viruses, analysis of a larger number of nucleotide sequences should be performed as they become available (
4).
CONCLUSION
Thisisthefirstreport of isolationofclass IAAvV- 1 in the Southeastern Europe. The asymptomatic nature of class I viruses results in the significant gap of available sequences and detections globally, thus impeding the understanding of the molecular epidemiology and evolution of these group of viruses.
CONFLICT OF INTEREST
The authors declared that they have no potential conflict of interest with respect to the authorship and/or publication of this article.
ACKNOWLEDGEMENTS
The complete F gene sequence in this study has been generated through the Sequencing Services of the Animal Production and Health Sub-programme of the Joint FAO/IAEA Division in Vienna, Austria.

 10.2478/macvetrev-2019-0015
10.2478/macvetrev-2019-0015



