Abstract
This study provides insight into the development and validation of the simple, rapid and sensitive method of Liquid Chromatography-Tandem Mass Spectrometry for determining the residues of dexamethasone in bovine milk. The maximum residue limit for dexamethasone in bovine milk is 0.3 ng/ml-1. Chromatographic separation was achieved on an Agilent Proshell 120 SB:C18 (2.7μm 100x3.0 mm) column. The bovine milk was extracted with ethyl acetate, and the evaporated elution was dissolved with hexane, water and methanol, and analyzed by the LC-MS/MS method. The mobile phase A was a solution of 5 Mm ammonium formate in water, while the mobile phase B was 0.1% formic acid in acetonitrile. The calibration graphics were prepared within the range of 0.15–0.6 ng/ml−1, and a successful linearity was achieved (r ≥0.999). The limit of detection was 0.016 ng/ml−1. The decision limit (CCα) and detection capability (CCβ) were 0.34 and 0.38 ng/ml−1, respectively.
Keywords: dexamethasone, LC-MS/MS, milk
INTRODUCTION
Dexamethasone is a synthetic compound of glucocorticoid hydrocortisone, which has been used in human and veterinary medicine. It is either in free form (alcohol) or in the form of esters such as isonicotinate, phosphate, dimethylbutyrate and phenylproprionate. It is used in the treatment of metabolic and inflammatory diseases. Dexamethasone is generally administered through intramuscular or intravenous injections of 20-60 µg/kg in horses, cattle and pigs (
1). Synthetic glucocorticoids are used in infection treatment as supportive therapy in order to shorten the clinical symptoms period along with antimicrobial drugs. As these drugs do not lead to acute toxicity when administered up to 10 times higher or even more than the ordinary therapeutic range, they can be administered in high dosages. The illegal use of dexamethasone as a growth-promoting agent in cattle breeding constitutes a serious health risk for meat consumers (
2,
3). In order to protect the consumer, maximum residue limits (MRLs) have been set for dexamethasone by the EU in several biological matrices such as muscle, kidney, liver and milk from different species. The maximum residue limit for dexamethasone in bovine milk is 0.3 ng/ml-1 (
4).
While the analysis of dexamethasone residue used to be carried out through HPLC-UV, GC-MS was preferred over it shortly afterwards as it lacked specificity. However, the need for derivatization before the GC-MS analysis used to cause waste of time. Thanks to the improvements in the LC-MS/MS technology, it has become possible to analyze dexamethasone residues with reaction monitoring mode (MRM) in particular (
5,
6,
7).
MATERIAL AND METHODS
Standards and chemicals
Methanol LC-MS grade and ethyl acetate were obtained from AppliChem GmbH (Darmstadt, Germany). LC-MS grade Acetonitrile was purchased from Chem-Lab Nv (Zedelgem, Belgium). Ammonium formate was provided from Acros Organics (Geel, Belgium). N-hexane was obtained from VWR Prolabo (Fontenaysous-Bois, France). Dexamethasone and dexamethasone-4, 6 alpha,21,21 D4, which was used as internal standard (IS) was obtained from A2S (Saint Jean d'Illac, France). Stock solutions were prepared in MeOH in a concentration of 1000 µg/ml−1 and stored at − 20 °C. The dexamethasone stock solution prepared in methanol is known to remain stable for two months at +4 °C refrigerator temperature (
8). The milk samples which were spiked with dexamethasone at a rate of 0.3 ng/ml−1 and stored at −20 °C were analyzed 5, 10 and 15 days later, and no significant change has been detected in their recovery levels (
9). It has been shown that the temperature of the autosampler affects the analysis results. Therefore, it is advised that the autosampler temperature is set at +4 °C (
10). In this study, the standard working solutions (30 ng/ml- 1) were not tested for stability, and therefore, standard working solutions were freshly prepared on the day of analysis.
Equipment
Vortex mixer (Neuation, İndia), nitrogen evaporator (Biotage, Sweden), and laboratory centrifuge (The Thermo Fisher Scientific Inc., USA) were used for sample preparation. Chromatographic analysis was performed using HPLC (Agilent 1260; Agilent Technologies, Germany). The mass spectral analysis was performed on a 6460 triple quadrupole mass spectrometer from Agilent Technologies (USA) equipped with an electrospray ionization (ESI) source interface operating in positive ion mode.
Sample preparation
Milk (10±0.01 g) was weighed into a 50 ml polypropylene centrifuge tube and 100 µl IS (100 ng/ml-1) is added to all samples. Samples used for the preparation of calibration curves were also spiked with appropriate volumes (50, 100, 150 and 200 µl) of the standard working solutions of dexamethasone (30 ng/ml- 1). The sample was vortex-mixed and left for 10 minutes. Then, 10 ml ethyl acetate was added. The sample was slowly mixed (10 min) and centrifuged (4000 x g, 10 min, + 4 °C), and 7 ml supernatant was transferred into a glass tube, evaporated to dryness at 40 °C under a stream of nitrogen. The extract was then re- dissolved in 1,5 ml of n-hexane and vortex-mixed (2000 rpm, 5 min) briefly. 0.6 ml of methanol and water (20:80) was added, and after being stirred on a multi-vortex (1500 rpm, 5 min), it was centrifuged (2500 x g, 10 min, -5 °C). A sample was collected from the phase sitting at the bottom with a disposable plastic syringes 2 cc, filtered through a 0.2 micron RC filter and transferred into a polypropylene vial. 50 µl of the liquid was injected into the LC-MS/MS system.
Liquid chromatography
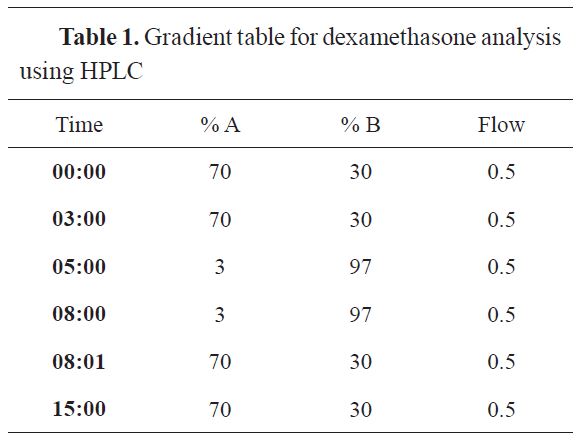
Chromatographic separation was performed on Agilent Proshell 120 SB:C18 (2,7µm 100x3,0 mm). The mobile phase A was a solution of 5 Mm ammonium formate in water, while the mobile phase B was 0.1 % Formic Acid in Acetonitrile. The column temperature was 40 °C, and the injection volume was 50 μl. The gradient table for dexamethasone analysis using HPLC is shown in
Table 1.
Mass spectrometry
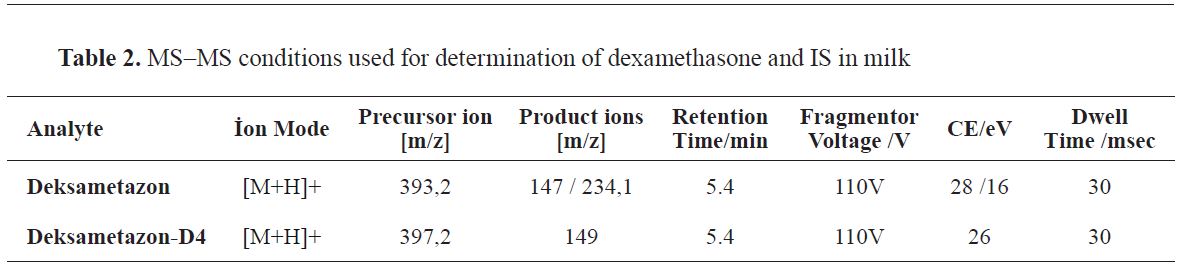
The analysis was performed using electrospray (positive mode), and the mass spectrometer was operated in multiple reaction monitoring (MRM) mode. After optimization, the source parameters were adjusted as follows: Gas Temp 325 °C, gas flow (l/min) 10, Nebulizer (psi) 45, Sheath Gas Heater 350, Sheat Gas Flow 10, Capillary Voltage 3500V. Two transitions of the analyte were monitored, one being for the internal standard. The MS MS values used for the determination of dexamethasone and IS in milk are shown in
Table 2.
RESULTS
Validation
Method Validation was performed according to the requirements of Commission Decision 2002/657 /EC (11). The parameters determined during validation are linearity, accuracy, selectivity and specificity, precision, decision limit (CCα), detection capability (CCβ), limit of detection (LOD) and limit of quantitation (LOQ).
Matrix matched linearity and range
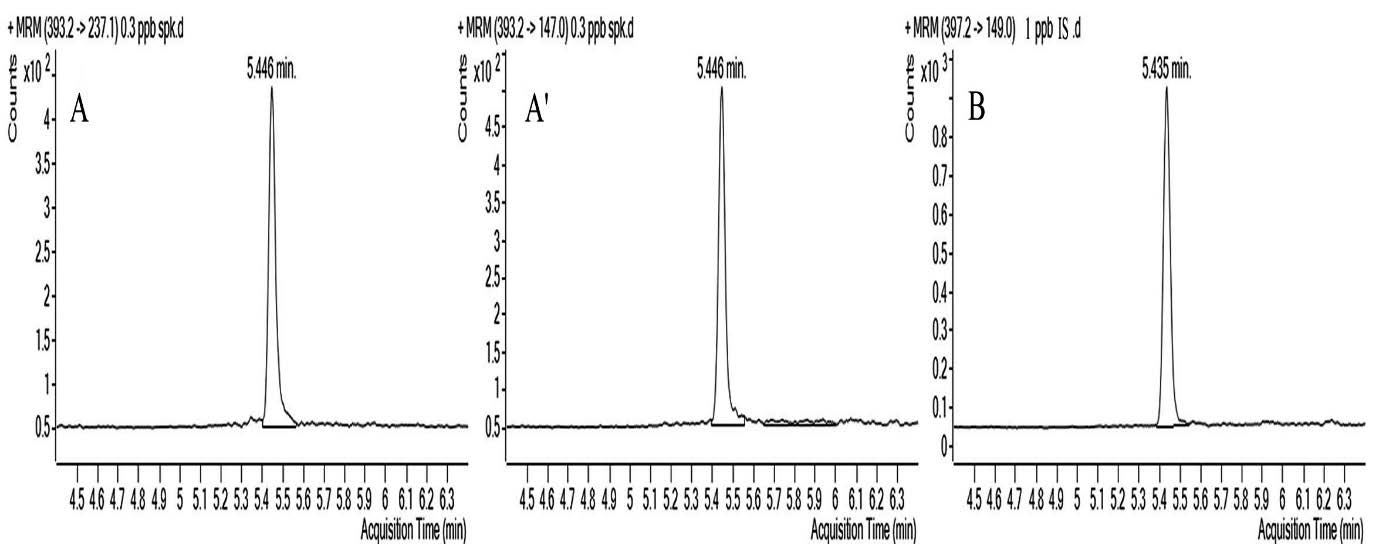
In an attempt to determine the linear measurement range, a blank sample and 4 samples with differing concentrations (0.15, 0.3, 0.45, 0.6 ng/ml−1) were prepared, calibration curves were drawn with the results acquired in three consecutive days. A successful linearity was achieved in the calibration curves prepared within the range of 0.150.6 ng/ml−1 (r ≥ 0.999). MRM chromatograms of bovine milk spiked with Dexamethasone are shown in
Fig. 1.
Specificity
The specificity was evaluated by analysing 20 blank milk samples. No interferences were detected in analyte retention time windows.
Decision limit (CCα) and detection capability (CCβ)
The CCα were calculated with the application of the following formula:
CCα= SDwlR,MRL*1,64+MRL
SDwMRL is the standard deviation from within-laboratory reproducibility (at the MRL);
The CCβ value can be calculated from the CCα value by the following formula:
CCβ= CCα + SDwlR,MRL*1,64
where SDwIR,MRL is the standard deviation of the MRL concentration.
Recovery
According to the decision of the European Commission Communities 2002/657 / EC, an acceptable recovery value of 1 µg/kg or less should be within the range of 50-120% (
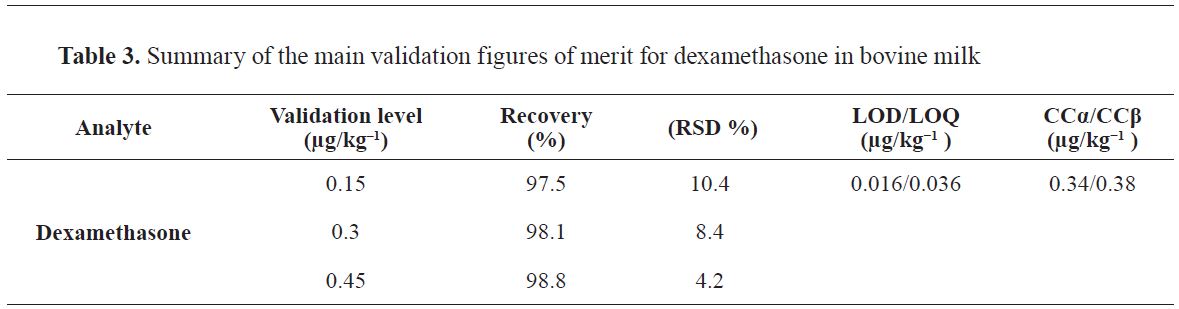
11). In the validation study, a level of less than 1 µg/kg was studied and it was observed that both analysts work with 50- 120% recovery in dexamethasone. Therefore, the validation study provides the recovery condition. Summary of the main validation figures of merit for dexamethasone in bovine milk is presented in
Table 3.
Figure 1. MRM chromatograms of bovine milk spiked with dexamethasone (A,A') (0.3 ng/ml−1), (B) dexamethasone-d4 (1 ng/ml−1)
DISCUSSION
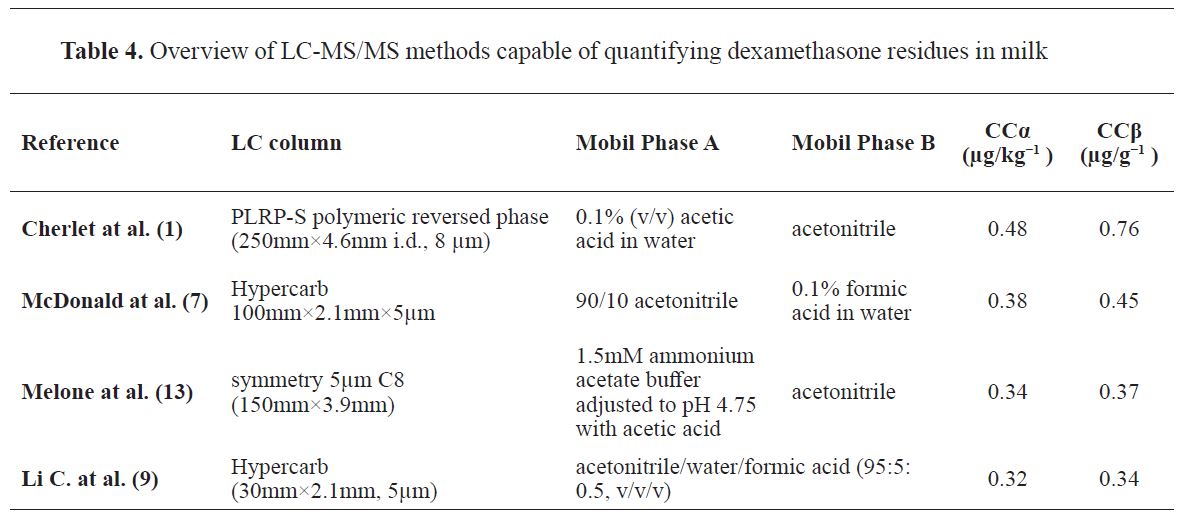
The aim of the control of veterinary drugs residues is to prevent that residues in the food will exert an harmful effect on humans. Indeed, the MRLs for the drug are based on the nonexistence of the this harmfull effects. Analytical methods for the determination of drug residues in food are established immunochemical, microbiological and physicochemical principles. Sample extraction is of essential importance when physicochemical techniques are used. The main objectives of sample extraction are removal of macromolecules and different matrix constituents that may either negative affect the chromatographic system, and the enrichment of the analytes in order to attain the necessary low limits of detection (
12). Many chromatographic methods (
Table 4) have been described in the literature for determination of dexamethasone residues in milk (
1,
7,
9,
13).
Since all current methodologies for dexamethasone residues determination use SPE as the extraction procedure, the development of a LLE method for sample extraction is of major value since it is not expensive by avoiding the use of SPE cartridges and faster. This method is proved to be simple, easy, and sufficient for high-throughput analysis at low cost.
CONCLUSION
This paper presents a simple and rapid LC-MS/MS method for determining dexamethasone in bovine milk. Among the published methods for detection of dexamethasone residues in bovine milk, there is not a published method for determining this level of concentration for dexamethasone without a detailed refinement procedure using a solid phase extracation. The results of this study indicate that this simple and rapid method can be applied to quantify dexamethasone residues below the MRL for raw bovine milk samples.
CONFLICT OF INTEREST
The authors declared that they have no potential conflict of interest with respect to the authorship and/or
publication of this article.
ACKNOWLEDGEMENTS
This work was funded by Pendik Veterinary Control Institute. The authors would like to thank the staff at Pendik Veterinary Control Institute, Department of Pharmacology, Laboratory of Residue for their practical assistance.

 10.2478/macvetrev-2020-0014
10.2478/macvetrev-2020-0014