The aim of this study was to construct two plasmid-specific shRNA transcripts of the bcl-2 gene in order to prepare for reverse of cell apoptosis. The plasmid was designed according to a previously published sequence of interfering RNA following an appropriate reference, using appropriate software. By annulling complementary oligonucleotides, doublestranded inserts were formed. Recombinant shRNA-encoding plasmids were constructed by digestion of psiRNAx7SKGFPzeo plasmid (psiRNA-x7SKGFPzeo, with restrictive endonuclease BbsI electrophoresis in ultra-pure agarose with low melting point (LMP-Agarose). For each of the constructs, a suitable double-stranded insert downstream of x7SK (strong RNA III promoter) with T4 DNA ligase was cloned. The control plasmid psiRNAScr was used directly for transformation. The PC-3 cell lines were transfected with 2 plasmids, psiRNA-Bcl-2 and psiRNAScr to suppress the bcl-2 gene construct. The results have shown that the lowest level of bcl-2 genes was 48 h, and even lower 72 h after the transfer, and the mRNA levels returned to normal in 120 h. An increase in the percentage of cells with spontaneous apoptosis has been observed with successful inhibition of the bcl-2 gene. The induction of apoptosis in transfected cells increased the percentage of necrotic cells proportionally. The percentage of apoptotic cells transfected with psiRNA-bcl-2 plasmid increased proportionally to the increase of hydrogen peroxide concentration. The transfection of the PC-3 cell line from prostate cancer with constructed shRNA plasmid has induced suppression of bcl-2 gene expression versus control Scr plasmid. Suppression of bcl-2 gene expression significantly increased cell sensitivity to apoptosis induction.
The basic traits of malignant cell are activating mutations in proto-oncogenes that cause continuous cell proliferation; inactivating mutations in tumor suppressor genes causing cells to be insensitive to anti-proliferative signals; activation of telomerase providing unlimited replication potential; neoangiogenesis (construction of new vascular network); ability to invade adjacent tissues, as well as to metastasize; and avoiding of the apoptosis due to overexpression of antiapoptotic or suppressed expression of proapototic molecules. All of these principles of pathogenesis are similar or the same in all studied types of human and animal malignancies of solid tissues. These different defense mechanisms are indicating the reasons why tumor changes occur relatively rarely during the lifetime. Apoptosis mediated by this pathway, is initiated on proapoptotic receptors located on the outer surface of the cellular membrane. They are activated by proapoptotic molecules or ligands specific to the appropriate receptor. These cellular death receptors are dependent on the group of tumor necrotic factor (TNF) receptors, represented by the Fas receptor and TNRF1, as the most studied members of this group. Fas is present in a variety of cell types, including activated B and T cells. Ligands that activate proapoptotic receptors include Fas ligands (FasL) and TNF. The intracellular DR protein domains are known as the Death Domain (
1, 2). After several decades of advanced research on tumors, it becomes clear that their complex basis lies in the dynamic disorders of the genome. Numerous studies have found a number of mutations that activate oncogenes with a dominant increase in their function, as well as genetic abnormalities that inactivate tumor suppressor genes by recessive loss of their function. These two classes of tumor genes have been identified in almost all human and animal malignancies and cause the characteristic tumor phenotype in experimental models (
19). Several findings indicate that tumorigenesis is a process that takes place through several steps that reflect genetic alterations that gradually lead to the progressive transformation of normal cells to malignant. Pathological analyzes on molecular level of numerous organs show the presence of lesions that occur as a result of processes that progress from normal to a series of pre-malignant to invasive forms of cancer (
3).
Prostate carcinoma is common malignancy in the male population, many of which with fatal ending (
3). Normal prostate epithelium is composed of luminal epithelial cells, basal cells, and a small fraction of neuroendocrine cells that adhere to the prostate (
4, 5). The majority of prostate carcinomas are adenocarcinomas, which are characterized by the absence of basal cells, and uncontrolled malignant cell proliferation, as well as luminal differentiation, including glandular formation and expression of androgen receptors (AR) and prostate specific antigens (PSA). It is an interesting fact that each mass of prostatic adenocarcinoma also contains a small population of non-tumor cells (~1%). These non-tumor cells have certain properties, found in benign tumors. For instance, unlike normal prostate cells, non-cells in benign tumors and prostate adenocarcinoma do not express AR and PSA (
6, 7). PC3 (PC-3) is a human prostate cancer cell line used in prostate cancer research and drug development. PC3 cells are useful in investigating biochemical changes in advanced prostate cancer cells and in assessing their response to chemotherapeutic agents (
7).
We hypothesized that hydrogen peroxide might induce apoptosis of the PC-3 cell lines of the prostate cancer. Therefore, this study aimed to investigate the sensitivity of PC-3 cell lines to hydrogen peroxide and to evaluate its effect on increasing the degree of apoptosis in PC-3 cells by suppressed expression of the bcl-2 gene. In addition, we investigated the effect of induction of apoptosis by treatment model with different concentrations of hydrogen peroxide (
8).
MATERIAL AND METHODS
In order to investigate the sensitivity of PC-3 cell lines of the prostate cancer to hydrogen peroxide, in general, several steps were undertaken. The first step was suppression of the bcl-2 gene in PC-3 cell cultures of prostate cancer. The second was establishing of a bcl-2 suppression technology through RNA interference mechanism as well as designing suitable recombinant plasmids encoding shRNA molecules, and production of sufficient quantity with the highest possible purity. The third step was evaluation of the ability to suppress bcl-2 using RNA interference to induce spontaneous apoptosis in PC-3 cell line prostate cancer and finally investigation of the sensitivity of PC-3 cell lines to hydrogen peroxide (
9).
Design, construction and control of the quality of plasmid constructsGenetic engineering techniques and general molecular-biology procedures were implemented according to the relevant laboratory manuals (
8), as well as according to the established methods in the laboratory. When commercial kits and products were used, the instructions and protocols recommended by the manufacturer were implemented (
9, 10).
Two plasmid-specific shRNA transcripts of the bcl-2 gene were constructed. The plasmid was designed according to a previously published sequence of interfering RNA, according to an appropriate reference, using appropriate software (siRNA Wizard, InvivoGen). Designed singlestranded DNA oligonucleotides with the highest degree of purity (PAGE) were ordered from a suitable manufacturer (SIGMA-Genosys). By annulling the complementary oligonucleotides, double-stranded inserts were formed. Recombinant shRNA-encoding plasmids were constructed by digestion of psiRNA-x7SKGFPzeo plasmid (psiRNA-x7SKGFPzeo, InvivoGen) with restrictive endonuclease BbsI (Fermentas International) containing an electrophoresis in ultra-pure agarose with low melting point (LMP-Agarose, SIGMA). An appropriate double-stranded insert downstream of x7CC (strong RNA III promoter) with T4 DNA ligase (Rapid Ligation Kit, Fermentas InteRNAtional) was cloned for each construct. In this manner, the control plasmid psiRNAScr (InvivoGen) was ready to use and was used directly for transformation (
11).
Bacterial transformations were performed with
E. coli strain GT116. (InvivoGen), which is a deletion mutant on SbcCD allele, and for which the manufacturer claims that is suitable for amplification of hairpin-containing plasmids. In addition, the standard bacterial strain JM109 (SIGMA) was used, resulting in high plasmid yields. Because shRNA insertion clones are lateral to the Lac-Z alpha peptide cassette, it was removed by cloning, so the discrimination of transformed bacteria by successfully recombinant psiRNA plasmids was done by white/blue screening (loss of alpha complementation) on solid RNA LB-based substrate with galacto-zidase inductor IPTG and substrate X-Gal. Antibiotic selection was performed with the antibiotic Zeocyn. Five colonies from each bacterial culture were tested by polymerase chain reaction (PCR) with thermosycler amplification (Perkin Elmer GeneAmp PCR System 2400) with plasmid-specific primers (InvivoGen). Three isolates from each colony were verified by sequencing both chains with a fluorescent putty Sanger Dioxidant Sequencing (Applied Biosystems) with appropriate sequencing equipment (Applied Biosystems Genetic Analyzer 310). The verified bacterial colonies, each from a different shRNAcoding recombinant plasmid and from the control psiRNAScr plasmid, were cultivated in a large volume of liquid medium TB (Terrific broth, InvivoGen).
Plasmidal DNA were purified from any bacterial culture by Endotoxin-free GeneElute Maxi-Prep Kit (SIGMA). DNA concentrations in each of the four psiRNA plasmid constructs and in the psiRNAScr control plasmid were determined by UV spectrophotometry, and the integrity and size of the plasmid DNA was determined by agarose gel electrophoresis and superspirated diatrile. When necessary, plasmids were re-purified by appropriate technique and using Endotoxin-removal solution (SIGMA).
Cellular lines and cellular culturesCell culture procedures were performed in accordance with generally accepted measures and according to laboratory manuals. Representative continuous cell lines from several human cancers were used for the purpose of this study. The cells were sub cultured in a suitable medium (RPMI 1640. MEM or DMEM) supplemented with 10% fetal calf serum (FBS), 1x non-essential amino acids, 2 mM L-glutamine, as well as a mixture of antibiotics and antifungals (all listed reagents by SIGMA) and then incubated at 37°C with an atmosphere saturated with moisture and 5% CO2 (Heraeus). Special T-25 Falcon vessels and 35-mm Petri dishes were used as well as all other plastic, sterile, apirogenic accessories certified for cell culture (Corning). The sub cultivation was done with trypsin-EDTA treatment to divide the cells into an appropriate number of new vessels and fresh medium. All procedures were performed aseptically in a laminar chamber with sterile equipment (
11).
TransfectionEach cell line was tested with three different transfection agents: the standard method of coprecipitation with calcium phosphate and with two commercial lipid-based agents: LyoVec and LipoGen, both by InvivoGen. LyoVec is a mixture of the phosphonolipids dimyristoleoyl phosphonomethyl trimethyl ammonium (DMTPA) and dioleoyl phosphatidylethanolamine (DOPE). LipoGen is a reagent with the properties of both, cationic and fusogenic lipids. During the optimization experiments, different DNA concentrations and/or different DNA relationships with the transfecting agent were examined, in accordance with the recommendations from the manufacturer (
11).
The efficacy of transfection with each transfection agent and each cell line was determined by fluorescence microscopy. The EGFP: Zeo tape in psiRNA-x7SKGFPzeo plasmid encodes an enhanced variant of the green fluorescent protein (originally isolated from the Aequore Victoria jellyfish). This protein absorbs blue light (max. About 480 nm) and emits fluorescent green light (max. About 505 nm), allowing easy detection of efficiently transferred cells as well as non-fluorescent cells. By counting them under fluorescence microscopy (Humanscope, Germany), transfection efficiency was determined, expressed as the percentage of transfected cells (
12, 13).
For direct evaluation of the effects of RNAimediated suppression, the cells were tested immediately after selection with subcutaneous doses of Zeocyn as a method of transient enrichment of efficiently transfected cells. Short-term treatment with a selective antibiotic will be lethal to non-transfected cells and thus increase the number of successfully transfected cells, allowing the suppressive effect to be monitored. In an attempt to prepare cells with stable knockdown of gene expression, long-term antibiotic selection was performed in which stable transfectants (with a randomly integrated plasmid in the cell genome) were collected on cloning disks and pulsed. By gradual expansion of stably transfected clones, a large number of cells were obtained, necessary for the implementation of the additional changes in cell morphology, the ability to form colonies in semi-solid agar, and other traits (
14).
Selection of the most efficient shRNA-encoding plasmid constructThe effectiveness of bcl-2 gene suppression constructs was evaluated by preliminary experiments with RT-PCR for relative quantification of transcripts from each of the three genes. The most efficient plasmids were selected for each of the three genes and were used in further experiments. Gene expression was compared with the same cells transfected with the control psiRNAScr plasmid (
14).
RT-PCRSemiquantitative RT-PCR to assess bcl-2 gene expressionThe sequence of the used primers was:
F 5’-ATGTGTGTGGAGAGCGTCAAC-3’
R 5’-TGCCGGTTCAGGTACTCAGTC-3’ (
28)
The expected length of the cDNA amplificant was 83bp.
For integral control of GAPDH cDNA (
15), with an expected cDNA amplificant length of 450bp.
The following primers were used:
(K136) 5’-CTCAGACACCATGGGGAAGGTGA-3’
(K137) 5’-ATGATCTTGAGGCTGTTGTCATA-3’ (
15)
Densitometric analysis for comparison of the intensities of electrophoretic strips from bcl-2 specific and control GAPDHgen were performed digitally using Image ver. 1.46 software (
14).
Cytotoxicity testsMTT analyses or colorimetric tests (SIGMA) were performed according to established methods (
16, 17, 18). These assays measure the activity of mitochondrial dehydrogenases in living cells.while the tetrazolium ring on the substrate was cut out in order to generate colored formazan product (3- (4,5-Dimethylthiazol-2-yl) -2,5-diphenyltetrazolium bromide) which was photometered. The reaction was enhanced by addition of phenazine methosulfate (PMS) to the mixture. The increase or decrease in the cell number and the subsequent change in the generated formazan product are reflection of the degree of cytotoxicity of the test agent (
14).
The total number of cells and the percentage of viable cells were determined by chemocytometer and the Trypan blue vital staining (SIGMA).
DNA laddering epectrophoretic test for apoptosis detectionThe test is based on the detection of inter nucleosome DNA fragmentation that occurs during apoptosis as a result of endogenous endonucleases. These enzymes cause double-strand gaps in chromosomal DNA at 180 to 200 bp-multiple intervals. It generates a series of DNA fragments that can be visualized by agarose gel electrophoresis with the characteristic “ladder” appearance of DNA-laddering (
14).
Apoptosis detection by fluorescent microscopyApoptotic and necrotic cells can be distinguished from the viable cells by acridine orange and etidium bromide cell staining. This test is based on the ability of acridine orange to penetrate all cells and produce green fluorescence of the nuclei. Etidium bromide, enters the cells only when the integrity of the cell membrane is disrupted and causes the nuclei to stain red-orange. The fluorescence of etidium bromide is more intense and is dominant over the fluorescence of acridine orange. Hence, viable cells have a normal green stained nucleus, early apoptotic cells have intensely green stained nuclei with condensed or fragmented chromatin; late apoptotic cells have condensed and fragmented red-orange stained chromatin. The cells that die by direct necrosis have a structurally normal nucleus with orange fluorescence (
14).
An epifluorescence microscope (Humanscope, Germany) with a set of filters was used (excitation maximum 450 nm/30 nm bandpass, emission maximum 540 nm/30 nm bandpass). The records were kept by digital camera (Cannon) and the images were processed on Adobe Photoshop 7 software for MS Windows XP.
Statistical analysisThe Student’s t-test was used to determine the significance of the obtained data by SPSS statistical software for MS Windows. An ANOVA test was used to determine if there is a statistically significant difference between PC-3 cell lines transfected with psiRNA-Bcl-2 and PC-3 cell lines transfected with psiRNAScr to suppress the expression bcl-2 gene construct by testing for differences of means using variance.
RESULTS
Selection of plasmids with suppressor expression of bcl-2 geneThe PC-3 cell lines were transfected with 2 plasmids, psiRNA-Bcl-2 and psiRNAScr to suppress the bcl-2 gene construct. The plasmid psiRNAScr encodes a small hairpin RNA with a “random sequence” that does not suppress the expression of any known human gene. Based on the results obtained, the lowest level of bcl-2 genes was 48 h, and even lower 72 h after the transfer, and the mRNA levels returned to normal in 120 h.
The suppression of the bcl-2 gene is presented in
Fig. 1. while semiquantitative values of bcl-2 mRNA levels are shown in
Table 1 and
Fig. 2.
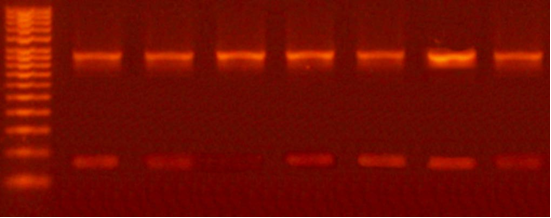 Figure 1.
Figure 1. Suppression of the bcl-2 gene determined by RT-PCR at 24, 48, 72, 96, 120, 144, and 168 hour intervals

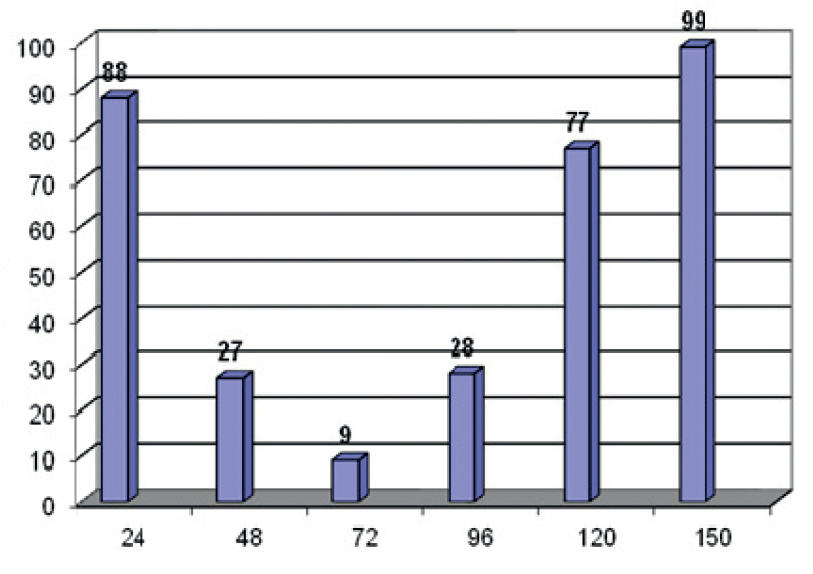 Figure 2.
Figure 2. Semiquantitative values of bcl-2 mRNA levels, measured by relative RT-PCR, expressed as percentages of levels in non-transferred cells from the PC-3 cell line
Levels of bcl-2 mRNAAs it can be seen from the above, the level of non-transferred PC-3 cells with bcl-2 mRNA is the highest in the first 24 hours, then it decreases, reaching the lowest level in 72 hours, and by 150 hours after the transection, there is a significant rise.
Levels of bcl-2 proteinQuantitative values of bcl-2 protein are given in
Table 2 and
Fig. 3.
It is notable that in PC-3 cell culture, the bcl-2 protein is on the lowest level in 96 hours, and the highest level is 150 hours after the transfection, which is slightly lower than the baseline.

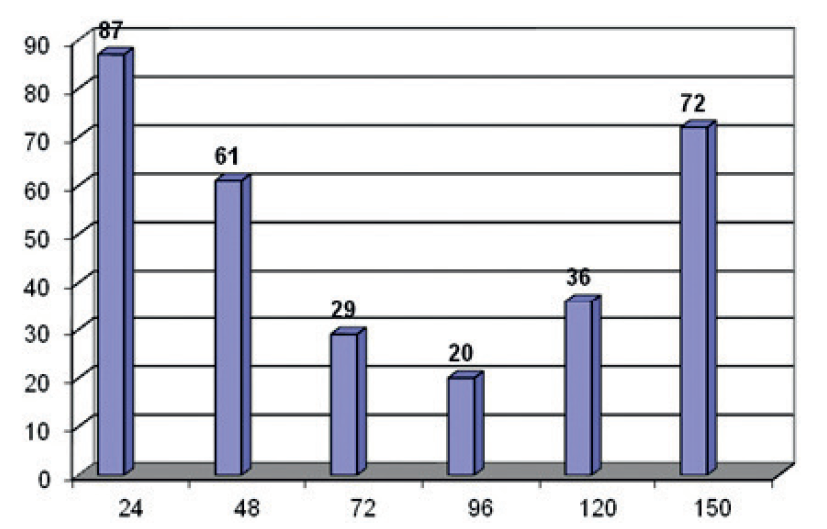 Figure 3.
Figure 3. Quantitative values of bcl-2 protein in a particular time dynamics
Treatment of PC-3 cell lines with hydrogen peroxideThe effect of hydrogen peroxide can be noticed. At 50μM/L there is a dramatic shift in the number of dead bcl-2 cells which is still one half less than the living ones. Later that proportion decreases, and at 200μM/L hydrogen peroxide, the increase in dead cells is twice as large as those that survive.
PC-3 cells- PsiRNA –ScrIt is evident that the number of dead cells increases in proportion to their sensitivity to increased concentrations of hydrogen peroxide, that is, as the amount of hydrogen peroxide increases, so does the percentage of mortality in cell culture (
Table 3,
Fig. 4. and
Fig. 5)

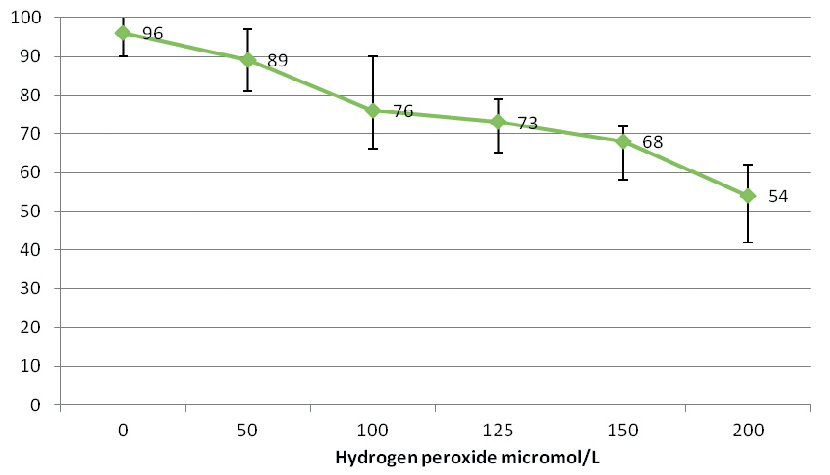 Figure 4.
Figure 4. The effect of hydrogen peroxide on transferred PC-3 cells with psiRNAScr
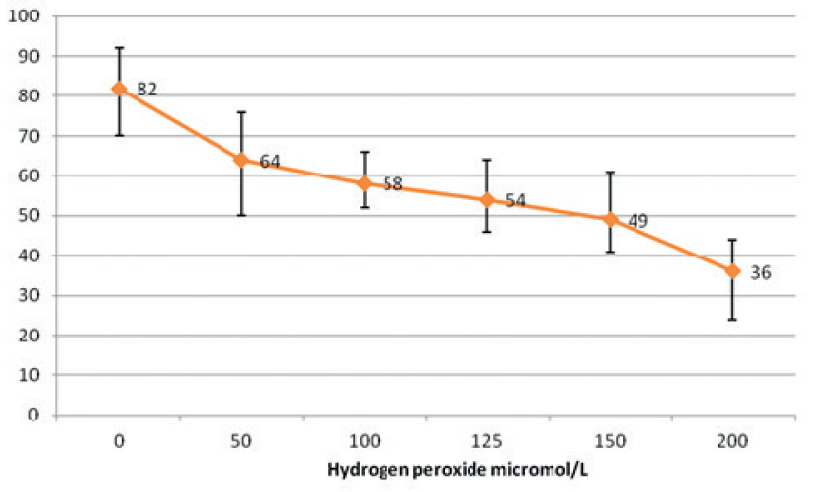 Figure 5.
Figure 5. Graphic representation of the effect of hydrogen peroxide on PC-3 cells transferred with psiRNA-bcl-2
PC-3 cells- PsiRNA –ScrThe sensitivity of PC-3 cells transferred with psiRNA-bcl-2 is slightly higher, so the level of dead cells after hydrogen peroxide treatment is higher.
Silencing of bcl-2 gene in PC-3 cellsStatistical analysis showed that PC-3 cell series transfected with Scr (control plasmid) and bcl-2 were statistically significant (p<0.05) using the one-way ANOVA (Tyrkes post-hoc test).
For apoptosis induction, the cells were treated with hydrogen peroxide, which showed major effect on the degree of apoptosis and necrosis in PC-3 cells. The change in the degree and ratio apoptotic: necrotic; viable cells mostly depended on the concentration of hydrogen peroxide solution, which ranged from 0, 50, 100, 125, 150 and 200 μM/L. At zero concentration of hydrogen peroxide in the culture of PC-3/psiRNA-bcl-2, the ratio of viable cells was 91.1%, apoptotic 6.8% and of necrotic only 2.1%, while in the control PC-3/Scr at the same concentration of hydrogen peroxide survival was 96.7%, apoptosis was found in 0.8%, and only 2.5% were necrotic (
Table 4 and
Fig. 6). When the concentration of hydrogen peroxide was increased at 50μM/L in the line PC-3/psiRNA-bcl-2 the ratio of was 66.1% viable, 19.7% apoptotic and 14.2% necrotic cells, which is different in comparison to the control PC-3/Scr line, where at the same concentration of hydrogen peroxide the ratio was 91% viable, 3.6% apoptotic and 5.4% necrotic cells. At hydrogen peroxide concentration of 100 μM/L in the PC-3/psiRNA-bcl-2 line the ratio was 58.7% viable, 51.3% apoptotic and 16.4% necrotic cells, in contrast to the control PC-3/Scr line, where, at the same concentration of hydrogen peroxide the ratio did not change significantly and was 71.3% viable, 12.9 apoptotic and 11.2 necrotic cells. When the concentration of hydrogen peroxide was 125 μM/L in the line PC-3/psiRNA-bcl-2 the obtained ratio was 32.3% viable, 33.5 apoptotic and 7.8 necrotic cells, which showed significant difference in comparison to the control PC-3/Scr line, at the same concentration of hydrogen peroxide, where the ratio was 75.9% viable, 26.4% apoptotic and 2.3% necrotic cells (
Fig. 7A, B and
Fig. 8 A, B). With the concentration of hydrogen peroxide at 150 μM/L, the effect on PC-3/psiRNA-bcl-2 increased dramatically compared to the lowest concentration and the ratio was 31.2% viable, of apoptotic cells were at 61.8% and the necrotic at 7%. In the control PC-3/Scr at the same concentration of hydrogen peroxide survival was 49.8%, apoptosis 29.9%, and only 20.3% of the cells were necrotic. The highest concentration of hydrogen peroxide at which the cell lines were treated was 200 μM/L, and the following results were obtained in the treatment of PC-3/psiRNA-bcl-2: 14.3% viable cells, 74.2% apoptotic and 11.5% necrotic cells, while in the treated PC-3/Scr, 51.3% were viable, only 34.6% were apoptotic and 14.1% were necrotic (
Table 5).
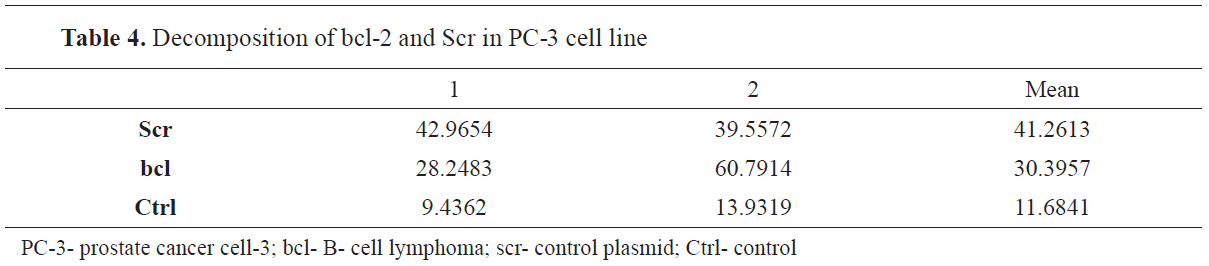
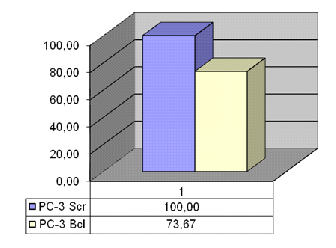 Figure 6.
Figure 6. Decomposition of bcl-2 and Scr in PC-3 cell line
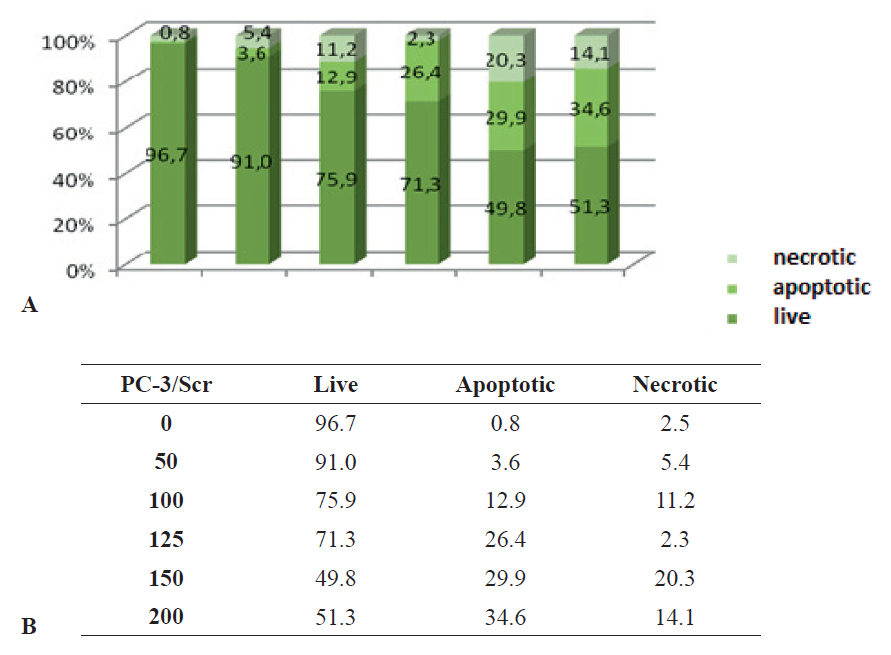 Figure 7 A, B.
Figure 7 A, B. Graphic representation of the percentage of apoptotic, necrotic, and live PC-3 cells transferred with Scr after treatment with different concentrations of hydrogen peroxide
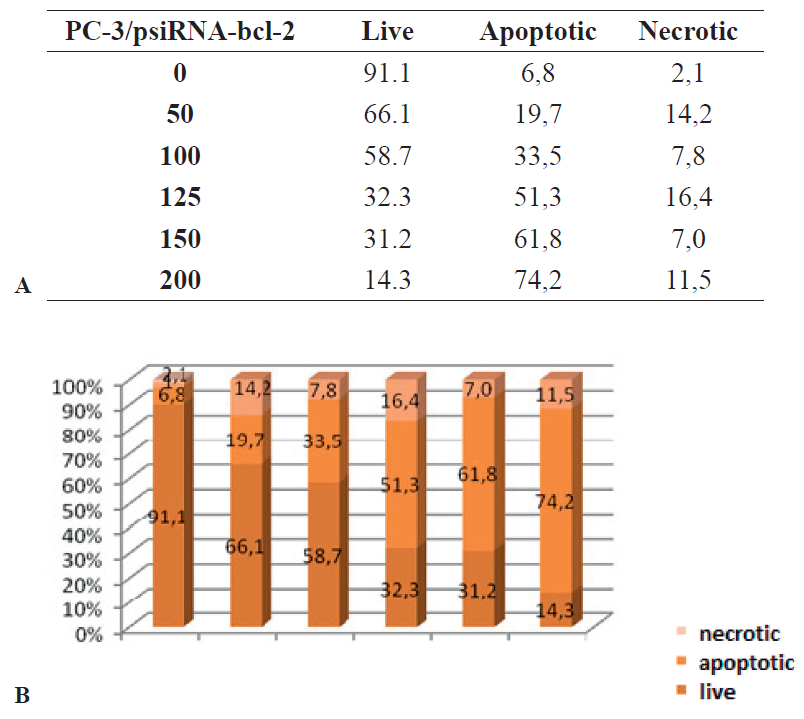 Figure 8 A, B.
Figure 8 A, B. Graphic representation of the percentage of apoptotic, necrotic, and live PC-3 cells transferred to bcl-2 after treatment with different concentrations of hydrogen peroxide

DISCUSSION
Malignant diseases are increasingly mentioned as the fastest growing mortality factor in the elderly population; however, the younger population is in an increasing risk, as well. The factors for the occurrence of malignant diseases are various. There are several theories for the occurrence of this disease, the most common of which are the viral theory, stress hormonal factors; the impact of the external environment and diet, as well as the evolutionary theory. Prostate cancer is a malignancy of one of the largest glands in men, including the testicles and seminal vesicles. When the tumor is growing, the cells are disseminated and metastasize to distant parts of the organism via the lymph and blood vessels. The most common targets of prostate metastases are lymph nodes, seminal vesicles, lungs, and the bones (
16, 17).
In this study, we focused on PC3 cells, which are used to study aggressive types of cancer, such as small cell neuroendocrine prostate cancer, which are also used to study androgen-dependent and castration-resistant tumors. Patients with small cell neuroendocrine prostate cancer often initially respond to aggressive chemotherapy treatment, but in a very short time they develop resistance to these types of therapies, significantly reducing therapeutic efficacy. The resistance of these tumors to different types of chemotherapy and radiotherapy leads to an excessive research aimed to change the genomic sequence of tumor cells and thus make them more accessible to commercially available types of cytostatic and radiotherapy (
18, 19).
If the resistant cell types are compared with the less resistant, the conclusion is that in the former, already reduced intracellular accumulation of the drug can be found, due to reduced activity of ATP-binding site of membrane pumps, reduced detoxification due to increased activity of glutathione-related enzymes, enhanced DNA repair of damaged parts of the molecule (
20) and systematic inhibition of apoptosis (
21, 22). Excessive research efforts were carried out in order to understand the real causes of such malignancies and striking conclusions emerged, pointing the complex nature of this disease: activation of proton oncogenes, inhibition of apoptosis, neoangiogenesis, immortalization, meta invasion and tissue invasion of tumor suppressor genes (
21).
Prostate malignancies are interesting in respect that they are closely related to the overexpression of the bcl-2 gene, and to the influence of the bcl-2 protein, which is directly involved in inhibition of the cell apoptosis and promoting the cellular “immortality.” Methods of destruction of bcl-2 proteins with small molecules are becoming increasingly important, and it is performed by designing small molecules that will be able to bind and inactivate the hydrophobic pocket of bcl-2 and Bcl-x, which is essential for the antiapoptotic function of the cell death promoter family (Bax, Bak, and Bad) through their BX3 domains (
23).
One of the most adequate biochemical specifics of apoptotic cell death is caspase activation. Once activated, these destructive proteases systematically destroy tumor cells without damaging healthy tissue. Caspase activity is the culmination of events and a bridge to the inhibitory mechanism that can develop in tumor cells to prevent a cascading reaction.
The common features of apoptosis are largely morphological and include chromatin condensation, DNA fragmentation, plasma membrane disruption, and cell destruction. In this study, a single cell line was used to suppress the bcl-2 gene and two recombinant plasmids, psiRNAScr (control) and psiRNA bcl-2, were constructed. The construction of the shRNA coding plasmid was done by molecular cloning. After transfusion in the cells, intracellular production of shRNA molecules is enabled, which are sliced by the Ditzer protein complex to form functional siRNA molecules (
24). The transfection procedure leads to inhibition of cell proliferation in psiRNAbcl-2, inducing short-term suppression of the growth of transferred cells, in contrast to the control psiRNAScr plasmid, which does not inhibit the expression of any known human gene. In addition, inhibition of the bcl-2 gene induced by plasmid transfection causes short-term inhibition of cell growth compared with psiRNAScr plasmid, which is transferred to the same PC-3 cells (
Fig. 9). In addition, according to the results from this study, the effects of bcl-2 gene suppression in PC-3 cells transfected with psiRNAbcl-2 are evident (
23).
An increase in the percentage of cells with spontaneous apoptosis has been observed with successful inhibition of the bcl-2 gene. The induction of apoptosis in transfected cells increased the percentage of necrotic cells proportionally. The percentage of apoptotic cells transfected with psiRNA-bcl-2 plasmid increased proportionally to the increase of hydrogen peroxide concentration. At a maximum H2O2 concentration of 200 μM/L, the viable cells transfused with psiRNA-bcl-2 were 36% and the dead cells were 64%, in comparison to cells transfected with the control plasmid psiRNA-Scr where the viable and dead cells were 54% and 46%, respectively. Suppression of bcl-2 gene expression after transfection with psiRNA-bcl-2 plasmid caused significant growth inhibition in the examined cells. These responses suggest that the effects of bcl-2 gene suppression are due to partial regeneration of cell proapoptotic pathways by suppression of the antiapoptotic role of the overexpressed bcl-2 gene in the cell line examined. Similar results were obtained after treatment of PC-3/psiRNAbcl- 2 cells with Docetaxel, which also inhibited microtubule formation, in addition to suppression of bcl-2 expression. Increased expression of bcl-2 is known to make cells resistant to radiotherapy. After treatment with docetaxel and bortezomib of the cell line with increased expression of bcl-2 it becomes sensitive to radiotherapy. Docetaxel and bortezomib have a pronounced cytotoxic effect resulting in the cessation of the G2M phase of PC-3/psiRNA-bcl-2. The cells treated in this way are much more sensitive to radiotherapy (
25). According to a different study on the effect of sodium butyrate (SB) on bcl-2 expression and the ability to induce apoptosis, similar findings for the effect of hydrogen peroxide on bcl-2 in PC cell culture 3 were obtained. Histone deacetylase inhibitors show anti-proliferative and apoptotic properties in a variety of tumor cells, including those of prostate cancer. The mechanism by which sodium butyrate (SB) induces apoptosis is not fully understood. The PC-3 cell line was treated in vitro with varying concentrations of SB. Condensed and fragmented nuclei were detected by analyzing the cell morphology with a laser scanning microscope. The conclusion was drawn that SB induces G1 and G2 phase arrest, as well as down regulation of bcl-2 and procaspase-3 in PC-3 cells. These results suggest that SB may serve as a model for the treatment of hormone-dependent prostate cancers (
24).
Expression of multiple shRNAs can inhibit multiple genes or multiple positions on the same gene. This is an important finding for establishing commercial genetic therapy protocols. An efficient shRNAs system based on lSUPER has already been established, which is also the most popular vector in mammalian cells. The multiple shRNAs vector is a set of 6 shRNAs fragments directed at specific genes involved in cell proliferation: bcl-2, Survivin, Akt1, Erk2, CyclinE, and NfkB. The inhibitory effects of multiple shRNAs expression vector on the human PC-3 line of prostate cancer, which contains different cell variants with different signal alterations, have also been evaluated. These findings point to the possibility of constructing short shRNAs fragments not only for bcl-2, but also for other, already known genes involved in cell proliferation and tumor survival. Multiple expression of shRNAs, in addition to inhibiting the aforementioned 6 genes, is effective in inducing apoptosis in PC-3 cells. These results suggest that a multi-targeted shRNAs expression system may be an effective strategy in cancer therapies, but also suggests for another DNA vector based on the shRNAs expression system (
26). Our research confirms the assumptions and results of similar research, which suggests the effect of hydrogen peroxide on PC-3 cell (
25, 26).
The invasive character of tumor cells plays a key role in metastasis, which is a major problem in the implementation of therapies. Urokinase plasminogen activator (uPA) and its receptor (uPAR) are major signal mediators for cell invasion, tumor survival, and metastasis of various types of tumors. For the purposes of the study, small shRNAs were constructed, targeting the human uPA and uPAR. These small interfering RNA constructs significantly inhibited the expression of uPA and uPAR, both at the mRNA level and at the protein level, resulting in decreased levels of mRNA and protein in the PC-3 prostate cancer cell line. As a result of such interference and reduction of the uPA-uPAR complex in PC-3 cell culture, a dramatic reduction in tumor cell invasion occurs. Simultaneous silencing of the uPA gene and its uPAR protein using a single plasmid expression construct shRNAs educates the cell capability and results in induction of apoptotic cell death (
27). In the analysis of shRNAs transfected cells for uPA and uPAR protein expression in PC-3 cells by semiquantitative reverse transcription, PCR revealed a specific decrease in mRNA levels (
27). RNA interference for uPA and uPAR interferes with the uPA-uPAR signal to target ERK/1 and Stat 3 molecules in the cascading process (
27). The results of another study suggest that intratumoral injection of a plasmid construct that expresses shRNAs for uPA and uPAR almost completely inhibits tumor growth and survival in a mouse model of prostate cancer. RNA interference that is directly targeted at uPA and uPAR is an easy and successful tool for studying the biological role of the uPA-uPAR complex, which will unequivocally increase the possibility of gene therapies utilization in the treatment of different types of cancer (
27).
CONCLUSION
Recombinant plasmids psiRNA-bcl-2 encoding shRNA molecules have been successfully constructed, targeting the bcl-2 gene transcript and post-transcriptional inhibition by the RNA interference mechanism. The transfection of the PC-3 cell line from prostate cancer with constructed shRNA plasmid has induced suppression of bcl- 2 gene expression. Suppression of bcl-2 gene expression significantly increased cell sensitivity to apoptosis induction. Overall, it can be concluded that the reduction of the expression of bcl-2 by RNA interference at the post-transcription level in cells having an overexpressed geneincreases the sensitivity of cells to induction of apoptosis, the rate of spontaneous apoptosis, the percentage of necrotic cells as well as it inhibits cell growth.
CONFLICT OF INTEREST
The authors declare that they have no potential conflict of interest with respect to the authorship and/or publication of this article.
ACKNOWLEDGMENTS
This research was supported by the Faculty of Veterinary Medicine-Skopje and Faculty of Natural Sciences and Mathematics.
AUTHORS’ CONTRIBUTION
SJ, NA and LM carried out of data samples and statistical data. LA,RC and AA isolated samples and established PCR protocols. IE mainly designed the study and supervised the whole program. NM and BA participated in experiment. IE and SJ wrote the manuscript. All authors contributed to revise the manuscript, read and approved the final manuscript.

 10.2478/macvetrev-2022-0028
10.2478/macvetrev-2022-0028



