Rabies is controlled by mass animal vaccination campaigns. Cats, dogs, and wild animals (e.g., bats) are large reservoirs of this virus and can pose a significant threat to the human health, especially in the developing countries. The nucleoprotein of the rabies virus is of great scientific interest since it has the potential to generate immunity in animals and can be used as for immunochemical diagnostics. The study aimed to test a large-scale expression of the rabies N protein in a prokaryotic system. The recombinant N protein was successfully expressed and purified. It was immunologically recognized by specific antibodies and was able to induce the production of specific antibodies in a mouse immunization assay. These encouraging results indicate that the recombinant N protein can be evaluated as an antigen for the development of a subunit vaccine or for a diagnostic assay.
Rabies is currently controlled through mass animal vaccination campaigns and reduction of the stray animal population through a program for the rescue and responsible adoption of stray dogs and cats (
2). Vaccination is achieved by means of different commercial vaccines developed for human and veterinary medicine.
Vaccines can be produced in nervous tissue and cell culture (
3). The production of biological subunit vaccines for rabies virus could be achieved under low biosafety level and could reduce the risk of infection for people who work with this highlydeadly virus (
4).
The rabies virus belongs to the genus Lyssavirus within the family
Rhabdoviridae. This virus has a negative-sense single-stranded ribonucleic acid (RNA) genome which encodes five proteins in the following order: nucleoprotein (N), phosphoprotein, matrix protein, glycoprotein (G), and RNA-dependent RNA polymerase (
5, 6).
The N protein of the Pasteur strain rabies virus contains 450 amino acids and has approximate size of 51 kDa (
7). It can generate immunity against rabies virus when administered to animals (
8). This protein is the main target for T-helper lymphocytes (
9) and has several characteristics that make it function as an exogenous superantigen (
10). The N protein is useful as viral antigen for immunochemical diagnostics of the rabies infection. Also, the antibodies produced by viral antigens have been used as diagnostic reagents to distinguish between rabies and rabies-related viruses (
11). The expression and cloning in
E. coli has a significant advantage in yields and purity (
12). The study aimed to test a large-scale expression of the rabies virus N protein in bacterial system for the potential use as antigen and/or immunogen design.
MATERIAL AND METHODSStrainsThe CVS strain of the rabies virus, provided by the Instituto Biológico Dr. Tomás Perón of La Plata, Buenos Aires, Argentina, was used for the extraction of viral RNA and for western blot assays. The pQE-30 UA vector (Qiagen, Germany) was used for cloning and expression of N protein in
E. coli strain M15 (Qiagen, Germany). This vector presents a Histag at the N-terminus to facilitate the purification of the proteins of interest. The Luria Bertani (LB) medium with 100 μg/ml of ampicillin and 25 μg/ml of kanamycin was used for the screening of
E. coli colonies transformed with the pQE-30 vector containing the N gene (pQE-30-N) and for the expression of the N protein in
E. coli (with Isopropyl ß-D-1-thiogalactopyranoside, IPTG, 1 mM).
Primer design and amplification of the N gene by RT-PCRTo amplify a segment of the N gene, a set of primers were designed from a multiple alignment with several sequences of the rabies virus obtained by BLAST (
https://blast.ncbi.nlm.nih.gov/Blast.cgi). In the design, the cleavage sites with the restriction enzymes
BamHI and
HindIII were added in forward (NF: GTCTCGAGGATCCATGGATGCCGACAA) and reverse (NR: CGGAAGCTTATGAGTCACTCGAATATG) primers, respectively. After RNA extraction of the CVS strain of rabies virus, the reverse transcription (RT) reaction was carried out with the reverse transcriptase enzyme of the Moloney Murine Leukemia virus (MMLV) (Promega, USA). The PCR reaction was carried out using cDNA, the NF and NR designed primers, and the high-fidelity enzyme Platinum SuperFi DNA Polymerase (Invitrogen, USA). The reaction was carried out under the following thermal cycling conditions: 94 °C for 3 min of initial denaturation; 94 °C for 45 s, 60 °C for 45 s, and 72 °C for 1.5 min (30 cycles); and a final extension of 10 min at 72 °C. In order to confirm the amplification of the N gene, an electrophoresis on a 1.5% agarose gel with 10 μl of the PCR product was carried out.
Ligation of the N gene in the pQE-30 vector and E. coli transformationThe
N gene and pQE-30 vector were digested with the restriction enzymes
BamHI and
HindIII (Promega, USA). The ligation was carried out following the protocol described for the ExpressLink ™ T4 DNA Ligase kit (Invitrogen, USA). This ligation mixture was used to transform electrocompetent bacteria (
E. coli M15) by electroporation. The transformation mix was plated on LB-Amp-Kan plates and incubated overnight at 37 °C. To confirm the presence of the N gene, 12 colonies were randomly selected and checked by colony-PCR with the NF and NR primers. The colony-PCR products were analyzed by 1.5% agarose gel electrophoresis. Finally, to verify the open reading frame of the cloned fragment, the plasmid DNA of one of the colony-PCR positive clones was purified and sequenced, using the sequencing primers included in the pQE-30 kit.
Expression of the N proteinWith the colony-PCR positive clones, cultures 1/100 diluted in 5 ml of LB-Amp-Kan medium were performed and incubated at 37 °C overnight, with stirring at 100 rpm until an optical density (OD)
600nm between 0.5 and 0.7. The cultures were induced with 1 mM IPTG for 2 h at 37 °C. Total proteins were extracted from each culture and the expression level of N protein was analyzed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western Blot (WB). A negative control from culture of M15 bacteria containing the empty pQE-30 vector was used. To optimize the concentration of IPTG and the induction time to generate the greatest amount of N protein, bacterial cultures were performed in 20 ml of LB-Amp-Kan with 0.5 and 1 mM of IPTG. From the beginning of the cultures, 2 ml were taken every hour (up to 3 h) from each flask. To compare the expression level of N protein of each culture, the total proteins of pellet samples were analyzed by SDS-PAGE and WB.
Rabbit anti-rabies polyclonal antibodiesFor N protein expression level assays by WB, we produced a rabbit anti-rabies polyclonal antibody by using the veterinary rabies vaccine provided by the Pasteur Institute of Buenos Aires, Argentina. The immunization protocol was carried out in a rabbit, which was immunized by subcutaneous injections with 1 ml of vaccine on days 0, 7, and 14. At 21 days post-inoculation, the rabbit was bled to obtain serum. As secondary antibody was used an antirabbit IgG (Sigma A9044, USA), conjugated with horseradish peroxidase.
Scale-up and purificationA batch culture was carried out in a 7.5-l BioFlo 310 Benchtop Bioreactor fermenter (New Brunswick-Scientific, Edison, NJ, USA), maintaining the flask conditions with the highest level of N protein expression. To perform the seed culture, 150 ml of fresh LB-Amp-Kan medium was inoculated with 1 ml of an overnight culture of the selected clone. After overnight incubation at 37 ° C with shaking, the culture was used to inoculate 3.85 l of new fresh LB-Amp-Kan medium in the fermenter (initial OD
600nm ~ 0.1). The temperature and pH were fixed at 37 °C and 7 °C respectively. The agitation was programmed to achieve aerobic growth of the microorganism and guarantee an oxygen concentration of at least 30% of the saturation value during all the process. Once the culture reached an OD
600nm of 0.6, it was induced with IPTG at a final concentration of 1 mM and kept for 3 h in culture. Culture samples were taken every hour, and, after 3 h, the culture was centrifuged at 3,000 x g for 20 min at 4 °C, discarding the supernatant. Finally, the pellet was analyzed by SDS-PAGE and WB. The purification was carried out by immobilized metal affinity chromatography in a 10 ml column loaded with a matrix of nitrilotriacetic acid and nickel (NTA-Ni) (Bio-Rad, USA), following the manufacturer’s recommendations. The eluted fraction containing the purified protein was dialyzed against phosphatebuffered saline, sterilized by filtration, and its concentration was estimated by means of an SDS-PAGE with a bovine serum albumin (BSA) standard and, using the TotalLab program.
Evaluation of the recombinant N proteinTo evaluate whether the recombinant N protein induce the production of specific antibodies against the same native protein of rabies virus, mice were immunized by the intramuscular (IM), subcutaneous (SC) and intraperitoneal routes (IP). After inoculation, serum samples were obtained from each animal and analyzed by WB. A single formulation containing 10% aluminum hydroxide as adjuvant (equivalent to 10 mg/ml aluminum content) and 10 μg/ml of the recombinant N protein was used. The veterinary rabies vaccine, produced by the Pasteur Institute of Buenos Aires in nervous tissue, was used as antigenicity control by the intraperitoneal (IP) route. A duplicate immunization protocol was carried out at 0 and 14 days by the intramuscular (IM), subcutaneous (SC), and IP routes. The amount of protein used in each route was determined according to previous reports (
13). At 28 days after the first inoculation, the mice were bled by cardiac puncture.
A WB was carried out with the CVS strain as antigen to analyze the presence of antibodies against the native N protein in serum samples. After the blotting and blocking steps, the polyvinylidene difluoride membrane was cut into thin strips and used individually with 1/50 dilutions of the serum from each mouse. After incubation for 90 min and three washes in washing solution, the membrane was incubated for 90 min with a 1/1,000 dilution of an anti-mouse IgG serum conjugated with horseradish peroxidase produced in goats (Sigma cat. A9040). Finally, after additional three washes, the presence of bound antibodies was detected using a solution containing 3,3’-diaminobenzidine (DAB) (Merk, Germany) and hydrogen peroxide.
Ethical committee evaluationThe experimental procedures developed in this study comply with international regulations (Institute of Laboratory Animal Resources, 1996) and were approved by the Institutional Committee for Care and Use of Laboratory Animals (CICUAL) of the Faculty of Veterinary Sciences of UNLP (Exp.600-004104/08-001. Res. 129/09).
RESULTSRT-PCR amplification
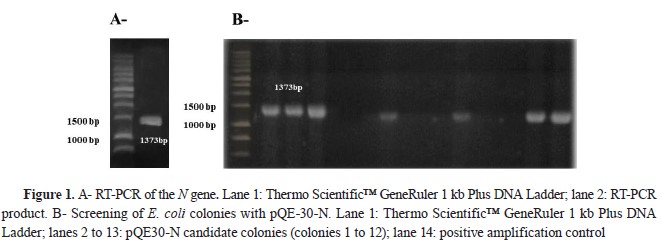
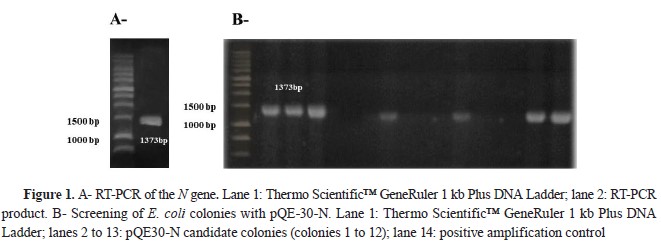
The amplification of the N gene was successful because an approximate size of 1.4 kb was observed (
Fig. 1A), which coincided with the size for the segment expected (1373 bp). Over 100 colonies were obtained after transforming
E. coli M15 cells with the pQE-30-N vector (N gene ligated to pQE-30 vector). Screening of the transformation of 12 colonies was carried out, showing a positive result in 6 colonies (1, 2, 3, 6, 9, and 12), confirming the pQE-30-N construction (
Fig. 1B). After sequencing, the start codon of the N gene was checked in frame with a His-tag in pQE 30-N. Also, analyzing the sequence by means of a local alignment by BLAST, more than 99 % sequence identity was obtained with the N gene of the viral strains Pasteur (GU992320.1), SAD-1 (EF206718.1), SAD-BERN (LN713619.1), and ERA (AB781935.1).
 Expression of recombinant N protein
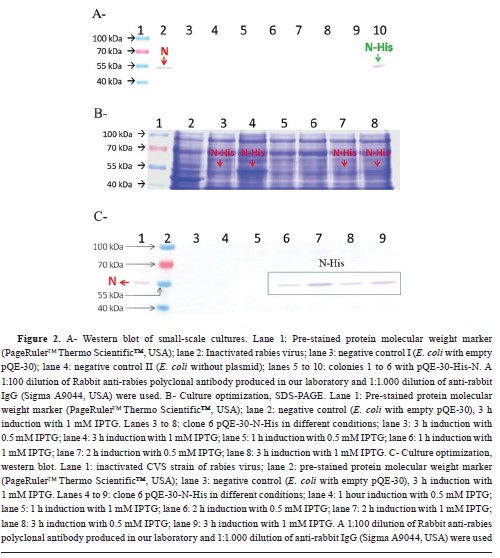
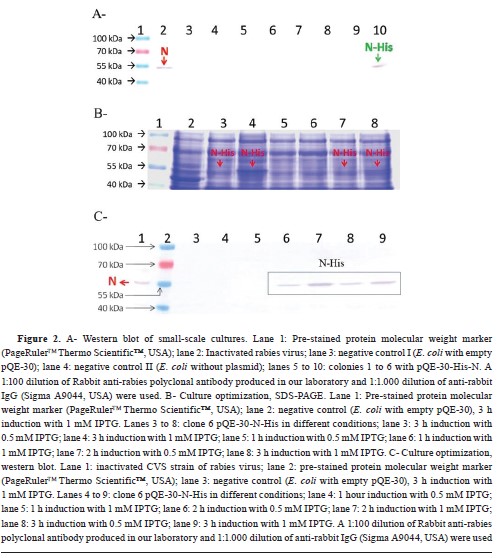
Expression of recombinant N proteinThe protein expression level assay by Western blot (WB) was conducted on the culture supernatant of the six colony-PCR positive clones (clones 1 to 6), as shown in
Fig. 2A. Among these clones, colony n° 6 exhibited recognition of His-N by specific antibodies against the rabies virus. Therefore, this clone was selected for further optimization of IPTG concentration and induction time required for scale-up. Subsequent analysis using SDS-PAGE (
Fig. 2B) demonstrated that the expression of His-N was highest when induced with 1 mM IPTG for a duration of 3 h. WB analysis (
Fig. 2C) was performed to confirm these results, and it revealed visible expression after a 2-hour induction period. Moreover, it was observed that higher levels of expression were achieved using 1 mM IPTG instead of 0.5 mM. Comparing the amount of His-N produced at both a 2-hour and a 3-hour induction period with the use of 1 mM IPTG showed greater expression at the latter time point. Based on these findings, it was determined that optimal conditions for scale-up involve using an IPTG concentration of 1 mM and inducing for a duration of 3 h. The purity and expression level of His-N in the scaled culture were qualitatively compared, observing them before and after purification (
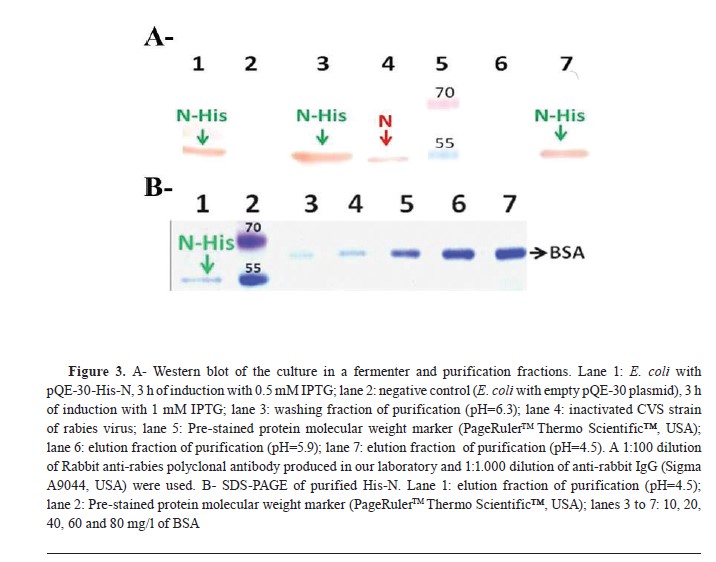
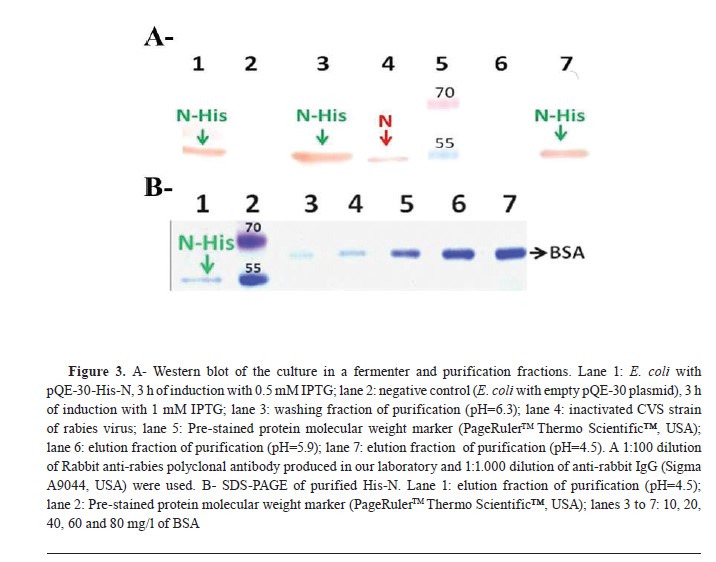
Fig. 3A). Furthermore, the concentration of N-His of the purified fraction was estimated at 30 mg/l (
Fig. 3B).
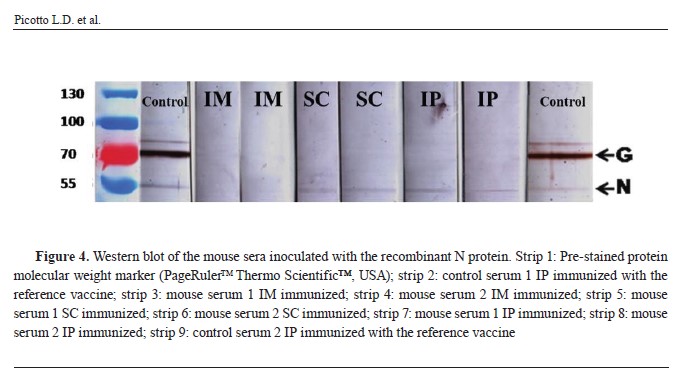
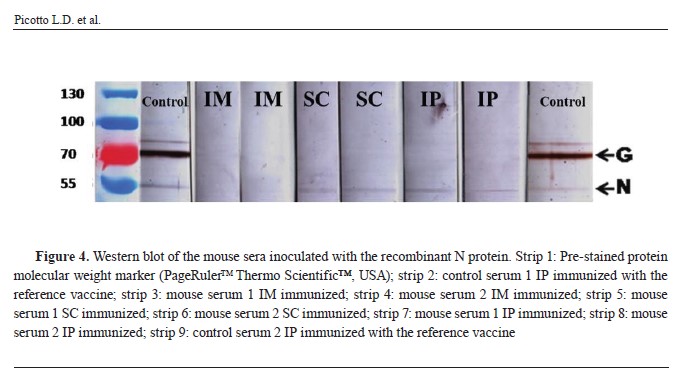
Immunological assaysThis qualitative test confirmed the presence of specific antibodies against native rabies N protein in the serum of the animals inoculated by the three routes with the recombinant N protein. The level of specific antibodies against native rabies N protein produced in the serum of the animals inoculated by the SC and IP routes was similar than that observed in animals immunized with the reference rabies vaccine used in Argentina (
Fig. 4).
 DISCUSSION
DISCUSSIONRabies is one of the oldest recorded diseases affecting humanity, dating back more than 2,000 years. Sporadic outbreaks of rabies in companion animals are increasingly frequent around the world (
14). This increases the risk of transmission to domestic animals, and consequently to humans, since they tend to interact with bats. The option to control the virus in affected areas is by a mass vaccination plan for animals that can act as viral vehicles and a reduction of the stray animal population. Adverse reactions may include neurological complications which are attributed to the myelinated tissue in the vaccine produced in suckling mice (
15), hence they have been discouraged by the WHO. Considering that rabies is a disease that mainly affects developing countries, an efficient and inexpensive solution is desirable. Recombinant or synthetic soluble antigens have been found to be safe and low-cost (
16).
This rabies virus genome encodes five proteins in the following order: N, P, M, G, and L (
5, 6). The most immunogenic and antigenic are N and G (
17, 18). The G-protein ectodomain stimulates neutralizing antibody production to protect vaccinated animals from rabies virus infection by inducing T lymphocytes and T helper cells (
9, 19, 20, 21). It has been extensively expressed in various heterologous systems, including yeast (
22). The N protein from different rhabdoviruses showed highly conserved characteristic with shared group-specific antigenic determinants (
23). Therefore, these proteins are great candidates to be used as antigens in diagnostic aims (
24) and/or as immunogen in triggering cellular immune responses against rabies (
10).
The aim of this work was to test large-scale expression of the rabies virus N protein in bacterial system with implications to be potentially used as a design for antigen and/or immunogen. The obtained recombinant
E. coli clones containing the N gene were subjected to a small-scale expression assay. Culture clones with the highest level of expression were selected. The IPTG concentration and the induction time to produce the highest level of N protein in flasks were optimized. The scale-up was carried out in a fermenter with the same conditions to improve the amount of N protein produced.
Plasmid stability is influenced by several factors such as copy number, the size of inserted DNA, and culture conditions, including temperature and growth rate (
25, 26). Higher growth rates are associated with increased plasmid stability, while cell density can impact the instability of expression plasmids during induction. Therefore, to address this issue, our strategy involved conducting IPTG induction at low cell density (in exponential phase) to prevent the loss of expression plasmids in a short time (
27).
The recombinant N protein produced and purified by affinity chromatography was specifically recognized by antibodies specific against the native N protein of the rabies virus. The recombinant N protein stimulated the production of specific antibodies against the native viral N protein in a similar level as the one reached the reference vaccine.
CONCLUSION
The recombinant N protein from rabies virus was produced in
E. coli and was purified by affinity chromatography. It was immunologically recognized and was able to produce specific antibodies against native rabies N protein in immunological assay. In combination with the recombinant glycoprotein, it can be used for development of subunit vaccine or a diagnostic kit.
CONFLICT OF INTERESTThe authors declare that they have no known conflict of interest in the conduction of the current study.
ACKNOWLEDGMENTSThis study was funded by National University of La Plata, Argentina “(Programa de Incentivos a Docentes Investigadores, Proyecto 11/V224)”.
AUTHORS’ CONTRIBUTIONLDP performed conceptualization, writing original draft, review and editing, conducted the investigation, methodology and visualization. CJP made the conceptualization, review and editing, visualization and formal analysis. MRP performed the review and editing. GHS conducted the investigation, review and editing of the paper.

 10.2478/macvetrev-2024-0026
10.2478/macvetrev-2024-0026