MATERIALS AND METHODS
Samples
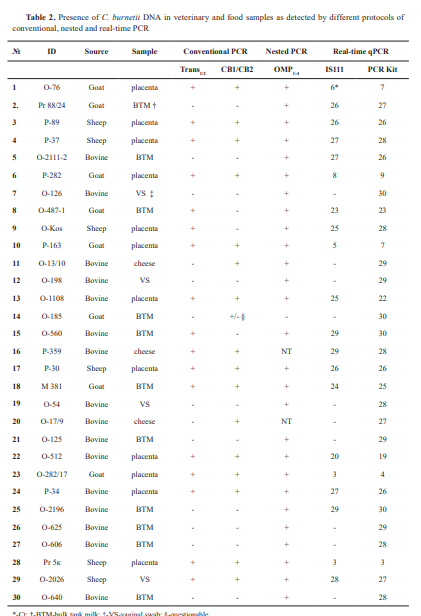
The study included archive genomic DNA extracted from samples scored positive for C. burnetii from previous diagnostic investigations, performed between 2014 and 2024, and retained for further analysis. These included 12 specimens from placentae, 4 vaginal swabs (VS) from cattle, sheep, and goats, 11 bulk tank milk (BTM) samples, and 3 cheese samples. They were analyzed by different conventional PCR protocols according to the primers available. From 2014 to 2020, PCR diagnostics in the laboratory was based on the use of primers CB1/CB2, targeting the superoxide dismutase (sod) gene of the Coxiella burnetii genome and/or Trans1/2 primers, comprising a sequence of the transposase gene of the IS1111 insertion element, while the OMP1-4 -PCR test was introduced later. Sixteen samples were of bovine origin, 8 were obtained from goats, and 6 were from sheep. Upon receiving the diagnostic materials in the laboratory, DNAs were isolated using commercial extraction kits, IndiSpin Pathogen Kit, (INDICAL Bioscience GmbH, Germany) for milk samples and PureLink Genomic DNA Mini Kit (Invitrogen) for tissue samples. After testing for the presence of Coxiella burnetii, extracted DNAs were stored at -20 °C for future reuse. This article does not contain any studies with human participants or animals performed by any of the authors. The research was conducted within the framework of a scientific project (Grant No KP-06-N33/3/2019) funded by the Bulgarian National Science Fund. The authors declare that they have the approval of the institution where they work to use the samples for research activities and to report the scientific results.
Conventional polymerase chain reaction (PCR)
The available DNAs were used as templates in different conventional PCR protocols. PCR reactions were conducted in a total volume of 25 μL. For Trans1/2 and CB1/CB2-PCR, the reaction mixture included 12.5 µL 2x Blank qPCR Master Mix (EURx Ltd, Poland), 2 µL of each primer at a concentration of 10 pmol µL-1, 2 µL target DNA and 6.5 µL nuclease-free H2 O (EURx Ltd, Poland). For the nested PCR with primers OMP1 -OMP2 and OMP3 -OMP4, respectively, the first amplification was performed in a total volume of 25 μL, containing 12.5 μL 2x Blank qPCR Master Mix, 5.5 μL nuclease-free H2 O, 1 μL primer OMP1, 1 μL primer OMP2, and 5 μL target DNA. For the second step, 2 µL amplification products from the first reaction were added to the reaction mixture consisting of 12.5 µL 2x Blank qPCR Master Mix, 8.5 µL nuclease-free H2 O, 1 µL primer OMP1 and 1 µL primer OMP2. The target genes for which the primers were designed, the oligonucleotide sequence, and the expected size of the amplification product are presented in Table 1.

All PCR reactions were performed in a DNA thermal cycler BIOER LifeECO at temperature regimes specific to each of the primer pairs used, as follows:
PCR with primers CB1/CB2. A modified procedure of Stein and Raoult (14) was used, with the amplification regimen consisting of an initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 54 °C for 1 min and elongation at 72 °C for 1 min and a final extension step at 72 °C for 10 min.
PCR with primers Trans1/2. Primers targeted a selected sequence of multicopy insertion element IS1111 in a modified PCR procedure of Berri et al. (16). It included an initial denaturation at 94 °C for 2 min followed by 40 cycles of 94 °C for 30 s, 61 °C for 30 s, and 72 °C for 1 min and final elongation at 72 °C for 5 min.
Nested PCR with primers OMP1 -OMP2 and OMP3 -OMP4 (15). The first amplification consisted of 94 °C for 4 min, 30 cycles of 94 °C for 1 min, 56 °C for 1 min and 72 °C for 1 min. In the second amplification, the temperature regime included denaturation at 95 °C for 4 min and 30 cycles of 94 °C for 1 min, 57 °C for 1 min and 72 °C for 1 min.
Тhe resulting PCR products were analyzed against DNA marker 100-1000 bp Hyper ladder, (Meridian Bioscience Inc.) or Gene Ruler 50 bp DNA Ladder, (Thermo Fisher Scientific Inc.) by electrophoresis in a 1.5 % agarose gel containing ethidium bromide and visualized in a UV transilluminator with a photo documentation system G:BOX (SYNGENE).
Real-time PCR
A bactotype C. burnetii PCR Kit (INDICAL Bioscience GmbH, Germany) was used according to the manufacturer's recommendations. Due to commercial considerations by the manufacturer, the target genes and oligonucleotide sequences were unknown. Negative and positive controls, as well as an internal (extraction/amplification) control were included in the kit.
In parallel, an in-house procedure of qPCR was developed to detect Coxiella burnetii by targeting a 154-bp long DNA fragment in the transposase of the insertion sequence (IS) element IS1111, using the primers CB_IS1111_0706F 5´-CAAGAAACGTATCGCTGTGGC-3´ and CB_IS1111_0706R 5´-CACAGAGCCACCGTATGAATC-3´ and probe CB_IS1111_0706P 6FAMCCGAGTTCGAAAACAATGAGGGGCTG-TAMRA (24). The in-house procedure consisted of the initial step of denaturation and polymerase activation at 95 °C for 10 min, followed by 40 successive cycles of denaturation at 95 °C for 10 s, hybridization/elongation at 60 °C for 30 s, and cooling at 40 °C for 30 s. DNA extracted from a Bulgarian C. burnetii strain, molecularly characterized by whole genomic sequencing (to be published), and retained in our laboratory, was used as a positive control. PCR-graded H2 O (EURx Ltd, Poland) served as a non-template negative control and was used in each PCR run. The real-time PCR reactions were performed on a CFX Opus 96 Bio-Rad Real-Time PCR system. All samples presenting Ct values lower than the positive control were considered positive. The assay was accepted to be valid when there was no signal in the negative control and the positive control yielded a fluorescence signal with a Ct<35.
Statistical analysis
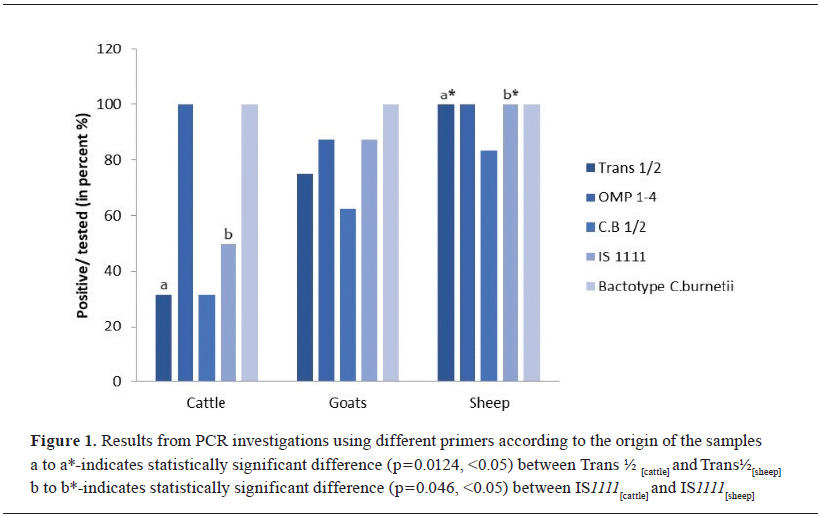
Data analysis was performed using the SPSS Statistics 26.0 (IBM Corp., Armonk, NY, USA) and Excel 2016 (Microsoft, Redmond, WA, USA) software platforms. A p-value of less than 0.05 was considered statistically significant. Chi-square test was used to test the association of tested positive samples with different primers based on 95% statistical significance. Fisher's exact test was used to determine the statistical significance of the differences between the groups.

 10.2478/macvetrev-2025-0020
10.2478/macvetrev-2025-0020